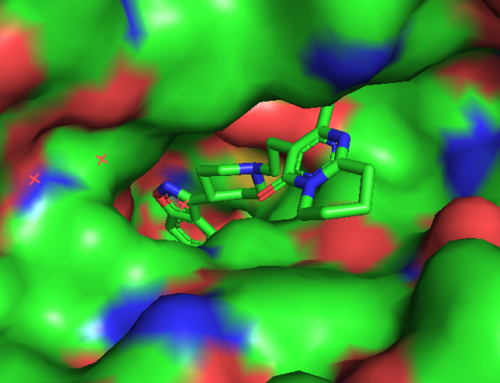
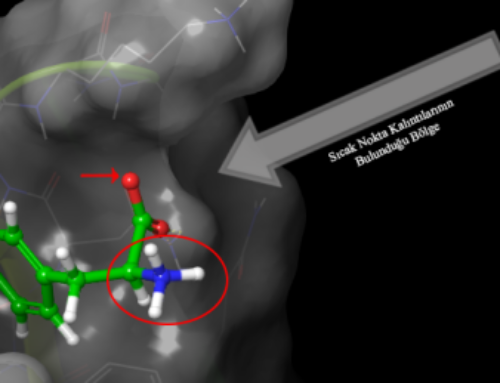
Moleküler Dinamik Nedir?
Moleküler dinamik parçacıkların hareketini incelemek için bilgisayar simülasyon yöntemleri kullanılmasıdır. Newton’un hareket denklemini sayısal olarak çözme bu hesaplamanın temelini oluşturmaktadır. Potansiyel enerji fonksiyonu ile yönlenen parçacıkların etkileşimleri (kuvvet alanı) hesaplamanın konusunu oluşturur.
Hesaplama işleminin sonucu, dış koşullara bağlı olarak sistemin belli bir zaman aralığında geçirdiği değişimin izlenmesidir. (e.g. pH 7, T = 300 K, p = 1 atm): trajectory
Moleküler Dinamik Simülasyonları Teorik Arka Plan
MD simülasyonu, biyolojik ve kimyasal makromoleküllerin dinamik davranışının teorik çalışmalarında ana araçlardan biridir. Bu bilgisayarlı hesaplama yöntemi ile bir biyomoleküler sistemin zamana bağlı davranışı hesaplanır. Atomlar, deney ve gözlemlere dayanan potansiyel enerji fonksiyonları veya çalışma alanına bağlı olarak seçilen çeşitli kuvvet alanları kullanılarak birbirleriyle etkileşim içine girebilirler ve atomlara etki eden her kuvvetler, bu fonksiyonlara veya kuvvet alanlarına dayalı olarak belirli bir konfigürasyon için hesaplanır. Newton’un hareket denkleminin (aşağıda gösterilmiştir) entegrasyonuyla, sistemin belirli bir zaman aralığı boyunca ardışık bir konfigürasyonu elde edilir.
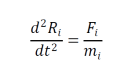
Burada Ri , i taneciğin konumunu temsil eder ve Fi diğer tüm moleküller tarafından uygulanan i parçacığına etki eden toplam kuvvet ve onun moleküler kütlesi mi ‘dir. Moleküler dinamik ile ilgili yapılmış farklı bir akademik bir çalışma American Chemical Society. Moleküler dinamik simülasyonlarında protein-ligand etkileşimleri için bir önceki adım yanaştırma yani docking işlemidir.
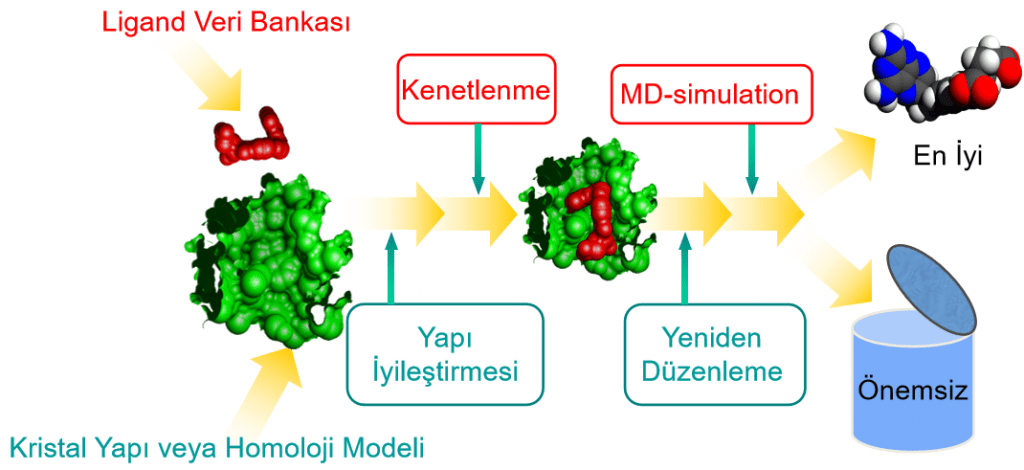
Moleküler Dinamik Kullanım Alanları ve Farklı Adımlar
Moleküler dinamik(MD) ilaç tasarımında protein ve ligand yapı etkileşimlerinin ilişkisini çözümlemede de kullanılmaktadır. Bir protein yapıya kenetlenen yani moleküler docking yapılan ligandın etkisi, bu yöntemle desteklenerek daha kesin sonuçlarla sunulur. Bu ilaç tasarımında, etkili ilaç adaylarının ayırt edilmesinde önemli bir adımdır. Bilgisayar destekli ilaç tasarımı yazısında ilaç keşfinin farklı adımları da anlatılmaktadır.
Kuvvet Alanı
Kimyasal bağların ve etkileşimlerin, enerji bazında hesaplanması için hesaplamalı kimyada matematiksel fonksiyonlar kullanılır. Hesaplamalı kimya ile analiz edilecek istemin potansiyel enerjisi, atomik konumların(koordinatların) bir fonksiyonu olarak kuvvet alanlarıyla tanımlanır. Şekil 3’de görüldüğü gibi, Moleküler Dinamik simülasyonları, bağların gerilmesini, bükülmesini ve ayrıca bağ yapmayan etkileşimleri içeren bir sistem içindeki etkileşimlerin deneysel olarak bulunmuş matematik fonksiyonlarına göre modellenmiştir.
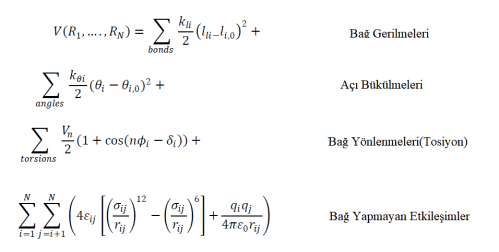
Yukarıdaki fonksiyonda, V(R1,….,RN ) potansiyel enerjiyi temsil etmektedir ve bu, N tane atom veya parçacığın koordinatlarının (Ri) şeklinde gösterildiği bir fonksiyonudur. Denklemde, ilk terim, bağ yapan atom çiftlerinin etkileşimini temsil eder ve li , bağ uzunluğunun ifade edildiği terimdir. İkinci terim Hooke Yasasın’nın matematiksel ifadesidir ve harmonik potansiyel olarak modellenen moleküldeki bağların açılarının toplamını ifade eder, burada Qi bağın ilk andaki açısını ve Qi,0 bağın denge açısını ifade eder. Denklemde, üçüncü terim burulma potansiyelini açıklar. Dördüncü denklem, iki farklı potansiyel ile temsil edilen bağ yapmayan etkileşimleri temsil eder. İlki, van der Waals etkileşimlerini açıklayan LennardJones potansiyel fonksiyonudur, ikincisi ise elektrostatik etkileşimler için Coulomb potansiyelidir.
Örnek Bir Çıktı
Protein yapıya kenetlenen ligand protein yapının hereketini engelliyor ise bu protein işlevini gerçekleştiremez veya normalden daha az gerçekleştirir hale geliyor demektir. Bu protein yapı bir reseptör veya enzin olabilir. Aşağıdaki videoda glutatyon redüktaz enziminin hareketi gösterilmektedir. Bunun yanında Raltegravir – İntegraz İnhibitörlerinin İlki adlı yazıda başka bir moleküler docking çalışması ayrıntılı olarak, bağlanma modlarını da içine alacak şekilde incelendi. Bir enzim inhibisyon mekanizmasının nasıl işlediğini anlamak için ideal yazılardan biri.
Bu görüntü, enzimin hareketi ile inhibitör bir ligandın kenetlendiği zamanki hareketini göstermektedir. Protein yapı ile iyi inhibisyon yapan bir ligand kenetlendiğinde protein yapı substrat ile yaptığı hareketinden daha farklı, daha “az” bir hareket yapacaktır. Bu MD simülasyonlarının değerlendirilmesi ve yorumlanmasında temel prensiplerden birini oluşturur.
MD’nin İlaç Geliştirme Çalışmalarımda Önemi
İlaç geliştirme çalışmaları temelde yapı bazlı ve ligand bazlı olmak üzere ikiye ayrılmaktadır. Özellikle yapı bazlı ilaç tasarımında bir enzimi ve ya reseptörü inhibe edecek ligandlar belirlenir. Yani sanal taraması yapılan kütüphanedeki ligandlar içerisinde en iyi inhibisyonu yapması öngörülen ligandlar. Bu ligandlar protein yapıya kenetlendikten sonra inhibisyon bölgesinde ne kadar süreyle durduğu ve protein yapının hareketinin ne ölçüde değiştiğine bakılır. En iyi kenetlenme puanına sahip ligandlar her zaman protein yapıya uzun süre kenetli olarak durmamaktadır. Bu durum sadece kenetlenme puanına bakarak bir ilaç keşfinin yapılamayacağı anlamına gelir.
Koronavirüs salgını ile mücadele ettiğimiz şu günlerde ilaç keşfi için gerekli olan çalışmalardan biri yine moleküler dinamik çalışmlarıdır. Covid-19 için ilaç keşif çalışmalarında favicovir ile ilgili pek çok veri bulunmaktadır. Favicovir ektili bir RNA proteaz inhibitörü olarak karşımıza çıkmakta. Favicovir ile ilgili daha çok bilgi için Favicovir Yapısı, Sentezi ve Özellikleri adlı yazıyı okuyabilirsiniz. Bu gün litaratürde yer alan etkili olma ihtimali yüksek ilaçların keşif ve yeniden amaçlandırma çalışmalarında moleküler dinamik simülasyonları da sıklıkla kullanılmakta.
Bir moleküler docking çalışmasıyla beş ayrı iyi inhibitör bulunduğunu varsayalım. Bu inhibitörler içerisinde birbirine çok yakın kenetlenme puanları elde eden ligandlar olabileceği gibi kenetlenme puanları bazında makasın açılması da söz konusu olabilir. Temel değerlendirmede bundan sonraki adım ligandların protein yapıda ne kadar süre ile kaldıkları ile ilgilidir. Kenetlenme puanı yüksek olan ligandların, protein yapıya kenetli kalma süreleri genellikle daha uzundur. Ancak kenetlenme puanı düşük olan ligandların protein yapıya daha uzun süre kenetli kalma ihtimalleri her zaman vardır. Zaten moleküler dinamik çalışmalarının yapılmasındaki temel nedenlerden biri de budur.
Moleküler Dinamik Verileri ve Yorumlama
Moleküler dinamik verilerinin yorumlanması için iki temel veri esas alınır. Bunlar RMSD ve RMSF grafikleridir. Bu grafiklerin haricinde pek çok veriye de ulaşılır. Bu veriler spesifik konularda araştırmacıya derinlemesine bilgiler vermektedirler. Bunların haricinde bir protein-ligand kenetlenmesinin değerlendirilmesinde RMSD ve RMSF değerleri genellikle yeterli bilgiyi araştırmacıya sunmaktadır. RMSD ve RMSF verilerinin haricinde alınabilecek diğer verilerin başında bağ türlerinin ve etkileşimlerinin zamanla değişimi(hidrofobik etkileşimler, hidrojen bağları ve bunun gibi), kenetlenen molekülün etkileşim içerisine girdiği amino asit kalıntılarının zamana göre değişimi, protein zincirindeki birincil ve ikincil yapıların hareketi başta gelmektedir. Aşağıdaki ligandın glutatyon redüktaz enzimine(PDB: 3DK8 DOI: 10.2210/pdb3DK8/pdb) kenetlenmiş halinin moleküler dinamik simülasyonu örnek olarak alınmıştır ve bu simülasyonun verileri anlatılmıştır.
Kök Ortalama Kare Sapması(RMSD) Nedir? Nasıl Yorumlanır?
Simülasyon yörüngesinin geri alınmasından sonra, protein (ve varsa ligand), zamana göre hareketlerini nicel olarak analiz etmek için sistemden izole edilir. Yörünge üzerinde yapılan ilk analiz, bir zaman adımı alınır ve atomlar hareket ederken, omurgadaki protein alfa-Karbon atomlarının başlangıç koordinatlarına göre kök ortalama kare sapmasıdır (RMSD). Bir ligandın bağlanması üzerine omurga C(alfa) atomlarının hareket serbestliğinde bir engel olması beklenir. Bu, enzimin bağlanma boşluğunun serbestçe hareket edemeyeceği, aktivitesini kaybedeceği ve işlevin engellendiği anlamına gelir.
Bu amaçla dimerik proteinin B zincirinin omurga C(alfa) atomlarının RMSD değerleri dışarıdan alınmış ve zamana karşı grafiğe geçirilmiştir. Bu adım, B zincirinin hem apo hem de holo formları için gerçekleştirilir(birden fazla zincir olması durumunda). Elde edilen grafikler, söz konusu zincirin enzimin hem apo hem de holo formlarındaki hareketini anlamaya yönelik netlik için tek alan üzerinde sunulur. Ligandın zamana göre hareketi de şekilde gösterilen grafiğe dahil edilmiştir. Aşağıdaki şekilde(grafik-1 ve grafik-2) gösterilen birimler, Ångstromlar olarak sunulmuştur ve değerler, proteinlerde veya ligandlarda farklı şekilde seçilen belirli atomların göreceli hareketinin gözlenmesine dayanak oluşturur. Proteinler için, kök ortalama kare sapmalarının hesaplanması için alfa Karbonlar seçilir. Ligand molekülleri için, omurga atomları genellikle aynı amaç için seçilir.
Hesaplamanın Özellikleri ve Hesaplama Süresi
Grafiklerin yorumlanması aşağıdaki örnek grafik ile yapılacaktır. Bu grafik 1,2 nano saniyelik bir hesaplama süresi ile çizilmiştir. Bunun anlamı ligandın protein yapıya kenetlendiği andan sonraki 1,2 nano saniye içerisinde neler olduğunun tüm atomlar bazında hesaplanması, hareketlerinin grafiğe geçirilmesi ve çeşitli etkileşimlerin gerçekleşme durumlarına ayrıntısıyla bakılması anlamına gelir. 1,2 nano saniyenin son derece kısa bir hesaplama aralığı olduğu unutulmamalıdır. Litaratürde 700 nano saniyeye kadar moleküler dinamik hesaplamaları yapılmaktadır. Bu veriler örnek olarak sunulmuştur. Hesaplama süresinin litaratür için geçerli bir süre olması elde edilen RMSD değerlerinin uygun bir plato oluşturması, yani belirli aralıklardan dışarı çıkmaması şeklinde ifade edilir. Bu belirli aralık iyi inhibe eden bir ligand için 3 Ångstromlar civarıdır ve olabildiğince 0’a yakın olması istenir. Bu olası plato seviyelerinin oluşması için gerekli hesaplama süresi genelde 50 ns civarlarıdır. Ancak hesaplama süresinin daha da uzun tutulması her zaman daha geçerli bir çalışma ortaya çıkmasını sağlayacaktır.
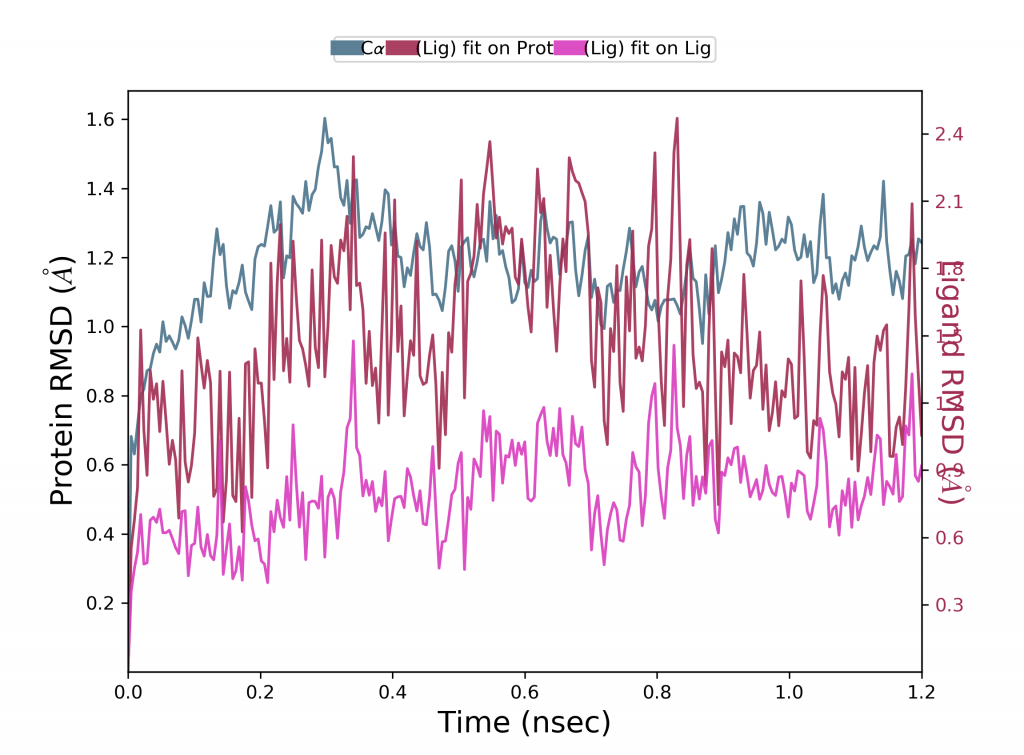
Protein RMSD nedir?
Yukarıdaki grafik, bir proteinin (sol Y ekseni) RMSD evrimini göstermektedir. Tüm protein çerçeveleri önce referans çerçeve omurgası üzerinde hizalanır ve ardından RMSD, atom seçimine göre hesaplanır. Proteinin RMSD’sini izlemek, simülasyon boyunca yapısal konformasyonuna ilişkin içgörü sağlayabilir. RMSD analizi, simülasyonun dengelenip dengelenmediğini gösterebilir. 1-3 Å sırasındaki değişiklikler, küçük, küresel proteinler için mükemmel şekilde kabul edilebilir. Bununla birlikte, bundan çok daha büyük değişiklikler, proteinin simülasyon sırasında büyük bir konformasyonel değişikliğe uğradığını gösterir. Simülasyonunuzun yakınsaması da önemlidir – RMSD değerleri sabit bir değer etrafında stabilize olur. Simülasyonun sonunda proteinin RMSD’si ortalama olarak hala artıyor veya azalıyorsa, sisteminiz dengelenmemiştir ve simülasyonunuz titiz analiz için yeterince uzun olmayabilir.
Ligand RMSD nedir?
Ligand RMSD (sağ Y ekseni), ligandın proteine ve bunun bağlanma cebine göre ne kadar stabil olduğunu gösterir. Yukarıdaki grafikte “Lig fit Prot”, protein-ligand kompleksi ilk olarak referansın protein omurgası üzerinde hizalandığında ve ardından ligand ağır atomlarının RMSD’si ölçüldüğünde bir ligandın RMSD’sini gösterir. Gözlenen değerler proteinin RMSD’sinden önemli ölçüde daha büyükse, ligandın başlangıçtaki bağlanma bölgesinden uzağa yayılmış olması muhtemeldir.
RMSD ve RMSF Değerlerinin Yorumlanması
Proteininin yapısındaki konformasyonel değişimlerin ligandın varlığı (holo form) ve yokluğunda (apo form) kıyaslanabilmesi RMSD ve RMSF değerlerini arasında kıyaslama yapılması verilerin değerlendirilmesindeki temeli oluşturur.
Grafik-1‘de açıkça gösterildiği gibi, enzimin omurgasının zamana karşı sapması ligandın sokulmasıyla engellenir. Apo formundaki omurga atomlarının ortalama RMSD değerleri 4,89 Å ve holo formunda 2,68 Å’dir. Bu, apo formundaki kalıntıların serbest hareketinin, ligandın bağlanması üzerine enzim boyunca inhibe edildiğini gösterir.
Kök Ortalama Kare Dalgalanması(RMSF) Nedir? Nasıl Yorumlanır?
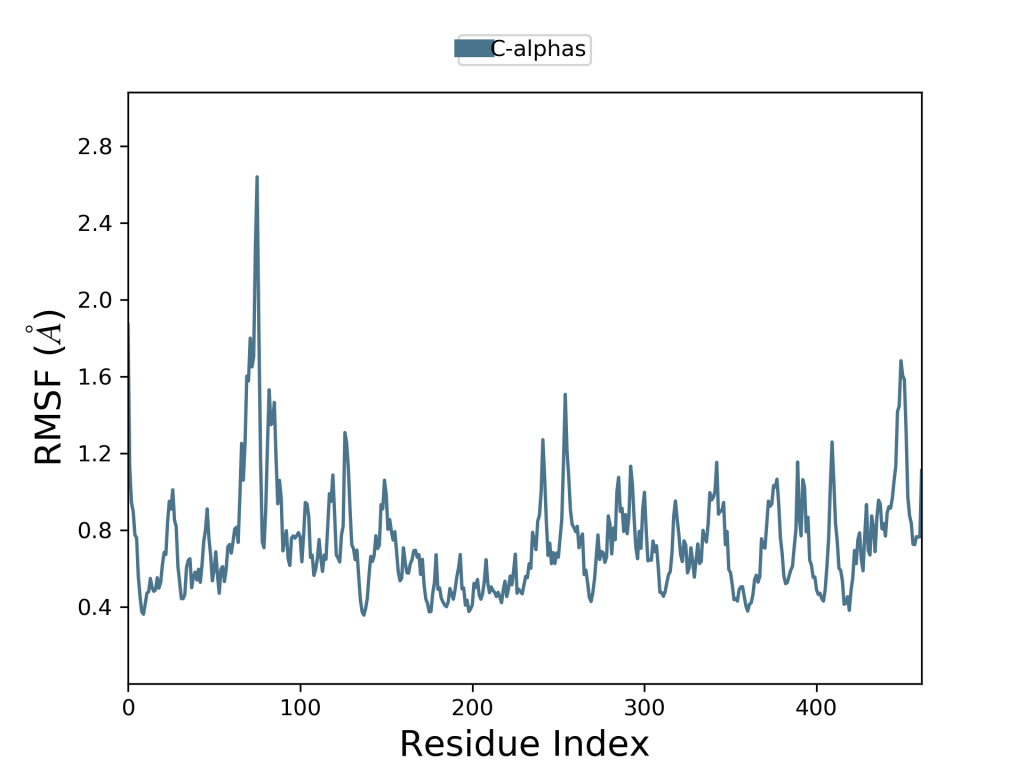
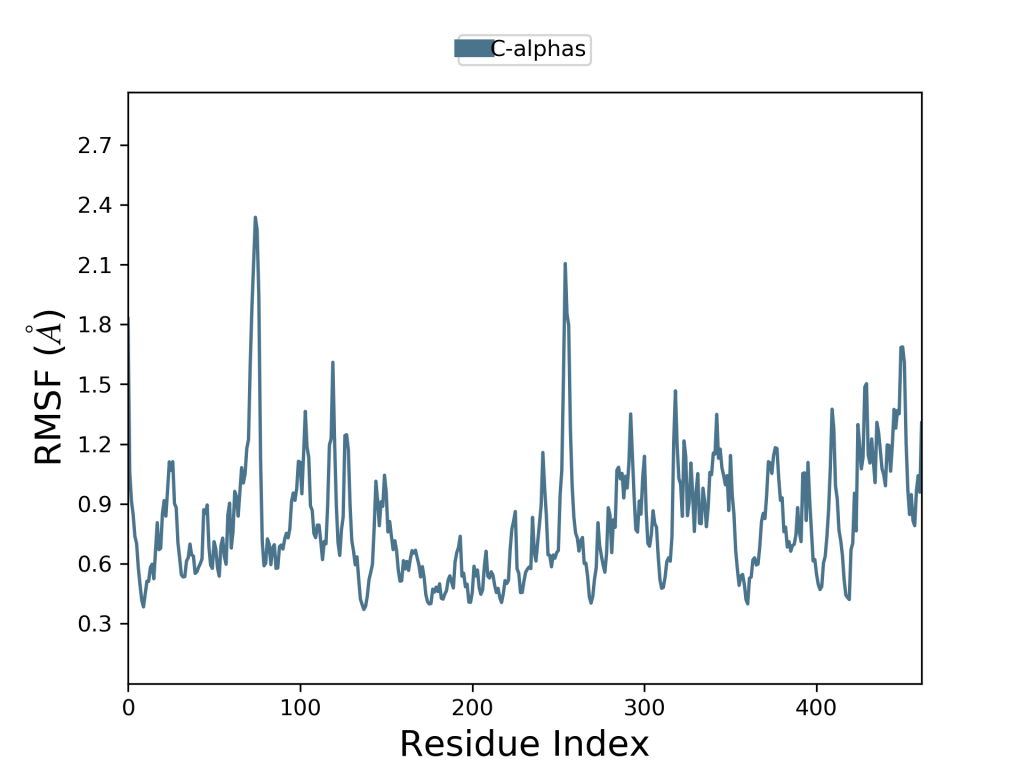
Yukarıdaki iki grafik (Grafik-2 ve Grafik-3) aynı protein yapıya kenetlenen iki ayrı ligandın RMSF grafiğidir. Bu iki grafiği bir biri ile karşılaştırarak hangi ligandın daha iyi inhihisyon yaptığını görmek mümkündür.
Moleküler dinamik simülasyonlarının ikinci analiz yöntemi, kök ortalama kare dalgalanması (RMSF) olarak adlandırılır ve dalgalanma hakkında bilgi verir. proteini oluşturan bireysel kalıntıların (veya hareketi). İki kök ortalama kare hesaplaması arasındaki fark, RMSF’nin bir zaman ortalaması olarak hesaplanması ve diğer yandan, RMSD’nin her zaman adımı için hesaplanmasıdır. İdeal bir MD simülasyonu durumunda, hedefin ikincil yapıları oluşturan kalıntıları (kalıntılar arasındaki hidrojen bağı etkileşimleriyle oluşturulan stabil yapılara sahip protein bölgeleridir), aktif bölgesinde bir inhibitör bulunduğunda daha az dalgalanır.
Örnek Analizi
Grafik-2 ve Grafik-3’ü karşılaştırdığımızda grafikte pik yapan yerlere bakmamız gerekmektedir. Moleküler dinamik simülasyonunda Grafik-3’ün pik yapan alanları açıkça daha fazla iken Grafik-2’de bu pik yapan bu alanlar azalmıştır. Bu da Grafik-2’deki ligandın, yani L1 ligandının, L4 ligandından daha iyi inhibisyon yaptığını gösterir. Bu yorumlama şekli protein yapının apo formunda da aynı şekilde kullanılabilir.
Bu grafiklere baktığımızda pik alanları ve plato alanları görülmekte bunun nedeni bu grafiklerde zirve olarak görünen yerler, simülasyon zaman aralığı içerisinde en çok dalgalanma gösteren protein alanlarını gösterir. Burada kuyruk kısımları olarak tabir edebileceğimiz (N- ve C-terminalleri de denilir) protein yapının diğer herhangi bir bölümünden daha fazla dalgalandığını gözlemlemek mümkündür. Alfa sarmal kısımları ve beta sarmal kısımları gibi ikincil yapı ögeleri genellikle proteinin yapılandırılmamış kısmından daha serttir ve bu nedenle döngü bölgelerinden daha az dalgalanır. Moleküler dinamik simülasyonlarının temel yorumlaması bu şekilde yapılmaktadır.
Moleküler Dinamik Simülasyonunda Diğer Veriler
Ligand ile protein etkileşimleri simülasyon boyunca izlenebilir. Bu etkileşimler, yukarıdaki grafikte gösterildiği gibi türe göre kategorize edilebilir ve özetlenebilir. Protein-ligand etkileşimleri (veya ‘temasları’) dört tipte kategorize edilir: Hidrojen Bağları, Hidrofobik, İyonik ve Su Köprüleri. Her etkileşim türü, ‘Simülasyon Etkileşim Şeması’ paneli aracılığıyla keşfedilebilecek daha spesifik alt türler içerir. Yığınlı çubuk grafikler, yörünge boyunca normalleştirilir: örneğin, 0,7 değeri, simülasyon süresinin% 70’inin spesifik etkileşimin korunduğunu gösterir. 1.0’ın üzerindeki değerler mümkündür, çünkü bazı protein artıkları, ligand ile aynı alt tipte birden fazla temas kurabilir.
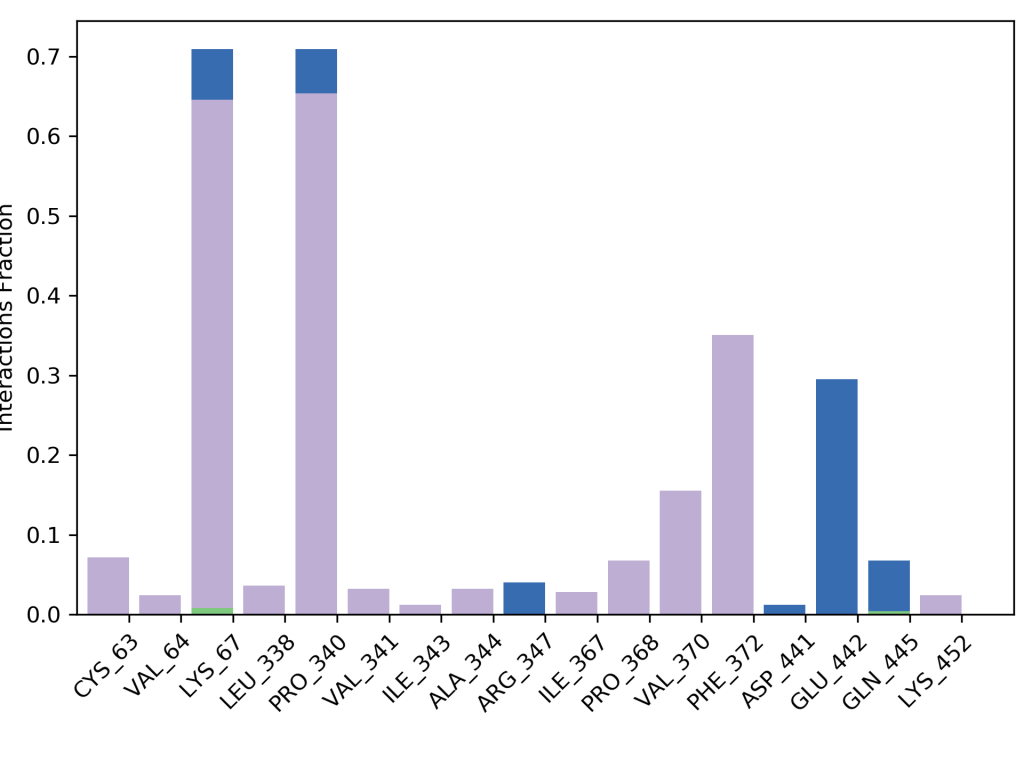
Amino Asitlerle İlişkinin Zamana Göre Değişimi
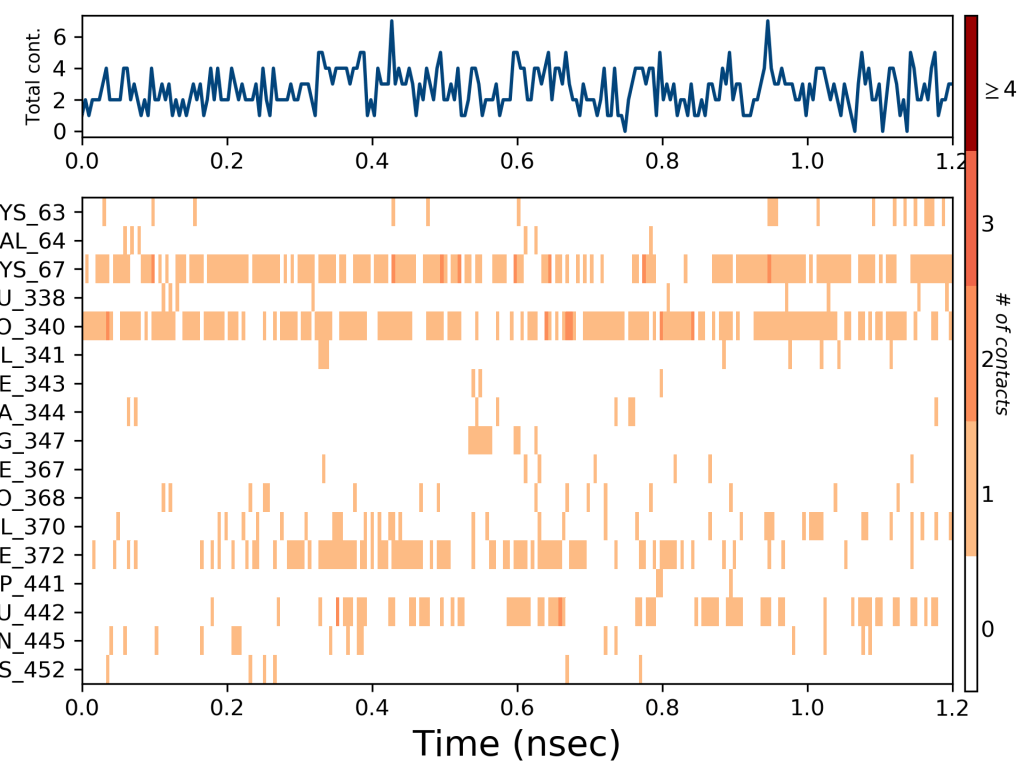
Yukarıdaki grafik glutatyon redüktaz enzimine kenetlenen yani moleküler docking çalışması yapılan bir ligandın hangi amino asit kalıntıları ile etkileşim yaptığını ve bu etkileşimlerin şiddetini göstermektedir. Grafikte görüldüğü gibi ligandımız en çok 67. lisin ve 340. prolin amino asit kalıntıları ile etkileşime girmiştir. Bu etkileşimlerin zamana bağlı değişimi de ayrıca gösterilmiştir. Yine grafikten açıkça görüleceği gibi 67. lisin ve 340. prolin amino asit kalıntıları simülasyon zamanı boyunca oldukça kuvvetli etkileşim içerisinde kalmıştır.
372. amino asit olan fenilalanin ile de zamana göre etkisi artan ve sonra azalan bir etkileşim söz konusu olduğu görülmektedir. 442. amino asit olan glutamin ile de yine zamanla değişen hafif bir etkileşim görülmektedir. Bu çalışmanın moleküler dinamik simülasyonundan sonra in vitro çalışması yapılmış ve yine L1 ligandının oldukça etkili bir inhibitör olduğu görülerek moleküler dinamik simülasyonlarının oldukça kesin bir şekilde doğru olduğu sonucuna varılmıştır. Glutatyon redüktazın in vitro çalışması için Glutatyon Redüktaz Enzim Aktivitesi yazımı okuyunuz. Tüm bu değerlendirmeler moleküler dinamik için RMSD ve RMSF değerlerinin yanında destekleyici veriler olarak görülmeli ve bütün olarak değerlendirilmelidir.
Torsiyon Açıları(Zamanla Değişim)
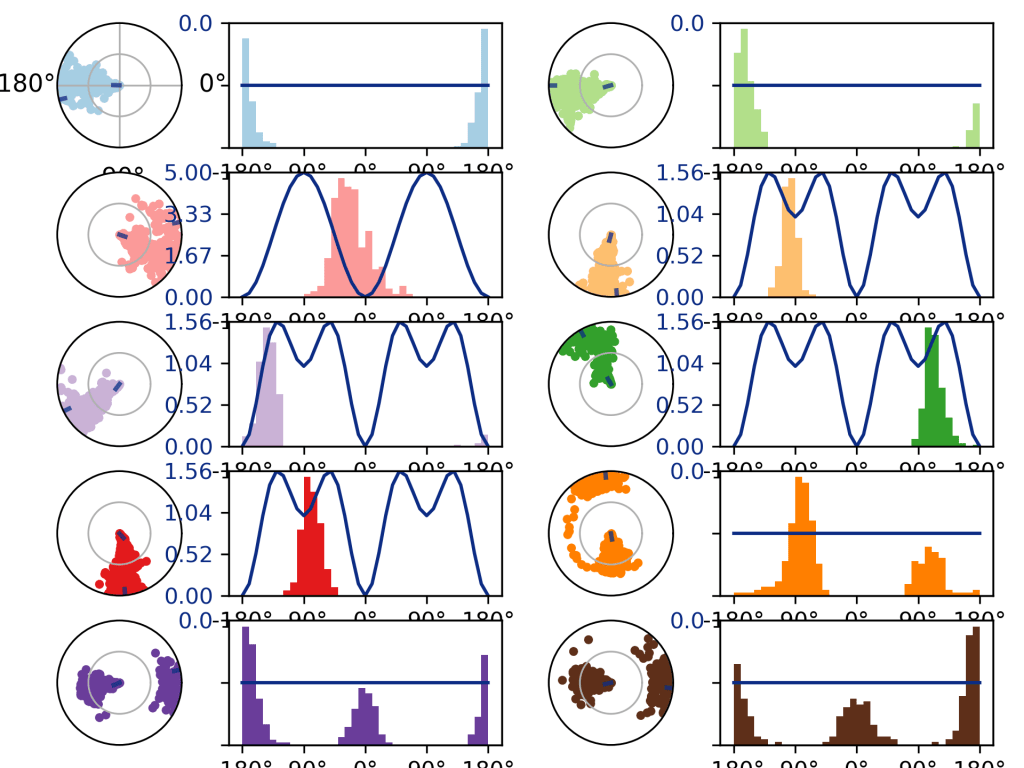
Ligandda yer alan, dönebilen yer bağ, protein kenetlenmesi ve sonrasında moleküler dinamik simülasyonları sırasında belirli açılarda döner. Bu da diğer veriler gibi simülasyon süresi boyunca izlenebilir. Yukarıda torsiyon açılarının hangi açılarda değiştiği yani simülasyon boyunca yönlenmenin miktarı açı cinsinden görülmektedir. Her renk ligandda bulunan bir bağı işaret etmektedir. Ligandın hangi açıya ne kadar zorlandığı(kenetlendiği protein yapının ve çözücünün etkisi ile) görülmekte ve grafiğe geçirilmektedir. Favicovir gibi koronavirüs için etkili olan ilaçlar da aynı şekilde yeniden amaçlandırma işlemi ve moleküler yakınlaştırma işlemine tabi tutulmaktadırlar. Akıllı malzemelerin nasıl elde edildiğini öğrenmek için Elastomer, Elektroreolojik Elastomer ve Özellikleri adlı yazıyı okuyabilirsiniz.