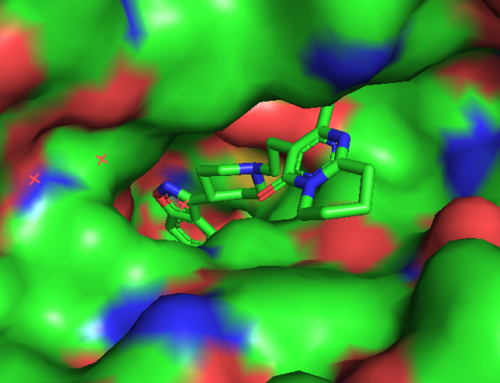
Lennard-Jones potansiyeli bağ yapmayan, yani aralarında belli bir uzaklık bulunan iki taneciğin birbiri ile etkileşiminin potansiyel enerjisini tanecikler arasındaki uzaklığı baz alarak ifade eder. Lennard-Jones potansiyeli moleküler dinamik simülasyonlarında en çok kullanılan potansiyeldir.
Moleküler dinamik simülasyonları, parçacıkların yani atomların hareketini incelemek için bilgisayar simülasyon yöntemleri kullanılmasıdır. Bu simülasyonlarda Lennard-Jones potansiyeli büyük önem taşır. Çünkü protein-ligand etkileşimler başta olmak üzere taneciklerin birbirine en çok ne kadar yaklaşacağının bilinmesi önemlidir. Moleküler dinamik simülasyonlarının diğer hesaplama alanlarını da içine alacak şekilde incelersek karşımıza; Newton’un hareket denkleminin sayısal olarak çözmesi moleküler dinamik simülasyonlarının temelini oluşturmaktadır. Potansiyel enerji fonksiyonu ile yönlenen parçacıkların etkileşimleri (kuvvet alanı) hesaplamanın konusunu oluşturur. Hesaplama işleminin sonucu, dış koşullara bağlı olarak sistemin belli bir zaman aralığında geçirdiği değişimin izlenmesidir.
Moleküler dinamik(MD) ilaç tasarımında protein ve ligand yapı etkileşimlerinin ilişkisini çözümlemede de kullanılmaktadır. Bir protein yapıya kenetlenen yani moleküler docking yapılan ligandın etkisi, bu yöntemle desteklenerek daha kesin sonuçlarla sunulur. Molek0ler docking yapılan yapı sadece bir enzim olmak zorunda değildir. Protein yapıların geneli için, protein yapının işlevsizleştirilmesi için bu tür çalışmalar söz konusu olabilir. Bu konuda daha ayrıntılı bilgi için Risin Proteini adlı yazıyı okuyabilirsiniz. Bu ilaç tasarımı konusunda, etkili ilaç adaylarının ayırt edilmesinde önemli bir adımdır. Bu konuda daha fazla bilgi edinmek için Docking ve Kanser Tedavisinde Kullanımı ve Yanaştırma Nedir? Moleküler Docking Nasıl Yapılır? adlı yazıları okuyabilirsiniz. Moleküler dockingden sonra ki adım olan moleküler dinamik simülasyonları ile ilgili daha fazla bilgi için Moleküler Dinamik Simülasyonları adlı yazıyı okuyabilirsiniz. Ayrıca akıllı malzemelerin tasarımı için Elastomer, Elektroreolojik Elastomer ve Özellikleri adlı yazıyı okuyabilirsiniz.
Lennard-Jones Potansiyeli Diğer MD Hesaplamalarıyla Birlikte Kullanılır
MD simülasyonu, biyolojik ve kimyasal makromoleküllerin dinamik davranışının teorik çalışmalarında ana araçlardan biridir. Bu bilgisayarlı hesaplama yöntemi ile bir biyomoleküler sistemin zamana bağlı davranışı hesaplanır. Atomlar, deney ve gözlemlere dayanan potansiyel enerji fonksiyonları veya çalışma alanına bağlı olarak seçilen çeşitli kuvvet alanları kullanılarak birbirleriyle etkileşim içine girebilirler ve atomlara etki eden her kuvvetler, bu fonksiyonlara veya kuvvet alanlarına dayalı olarak belirli bir konfigürasyon için hesaplanır. Newton’un hareket denkleminin (aşağıda gösterilmiştir) entegrasyonuyla, sistemin belirli bir zaman aralığı boyunca ardışık bir konfigürasyonu elde edilir.
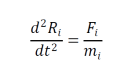
Burada Ri , i taneciğin konumunu temsil eder ve Fi diğer tüm moleküller tarafından uygulanan i parçacığına etki eden toplam kuvvet ve onun moleküler kütlesi mi ‘dir. Energy Landscapes of Quantum Lennard-Jones Solids adlı yazıda Lennard-Jones polansiyeli için pek çok hesaplama yapılmış veriler sunulmuştur.
Kuvvet Alanı Kavramı
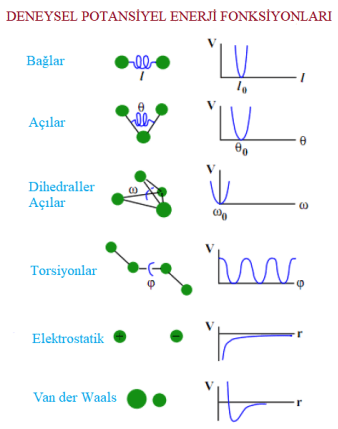
Kimyasal bağların ve etkileşimlerin, enerji bazında hesaplanması için hesaplamalı kimyada matematiksel fonksiyonlar kullanılır. Hesaplamalı kimya ile analiz edilecek istemin potansiyel enerjisi, atomik konumların(koordinatların) bir fonksiyonu olarak kuvvet alanlarıyla tanımlanır. Şekil 3’de görüldüğü gibi, Moleküler Dinamik simülasyonları, bağların gerilmesini, bükülmesini ve ayrıca bağ yapmayan etkileşimleri içeren bir sistem içindeki etkileşimlerin deneysel olarak bulunmuş matematik fonksiyonlarına göre modellenmiştir.
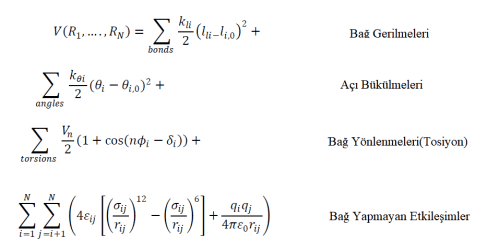
Yukarıdaki fonksiyonda, Ri(R1,….,Rn ) potansiyel enerjiyi temsil etmektedir ve bu, N tane atom veya parçacığın koordinatlarının (Ri) şeklinde gösterildiği bir fonksiyonudur. Denklemde, ilk terim, bağ yapan atom çiftlerinin etkileşimini temsil eder ve li , bağ uzunluğunun ifade edildiği terimdir. İkinci terim Hooke Yasasın’nın matematiksel ifadesidir ve harmonik potansiyel olarak modellenen moleküldeki bağların açılarının toplamını ifade eder, burada Q1 bağın ilk andaki açısını ve Q1,0 bağın denge açısını ifade eder. Denklemde, üçüncü terim burulma potansiyelini açıklar. Dördüncü denklem, iki farklı potansiyel ile temsil edilen bağ yapmayan etkileşimleri temsil eder. İlki, van der Waals etkileşimlerini açıklayan Lennard-Jones potansiyeli fonksiyonudur, ikincisi ise elektrostatik etkileşimler için Coulomb potansiyelidir. Moleküler dinamik için daha fazla teorik bilgiye https://doi.org/10.1021/acs.jcim.7b00674 linkinden ulabilirsiniz.
Lennard-Jones Potansiyeli Hesaplamarı için Sistemin Hazırlanması
Bir MD simülasyonunu gerçekleştirmek için sistemin hesaplamaya başlamadan hemen önceki zaman değeri sıfır olarak kabul edilir ve sistemin belirli bir zaman içerisindeki hareketi için hesaplama yapılır. Yani sistemin atomik koordinatlarını belirterek sistemin bir ön konfigürasyonu üzerinden hesaplama başlatılır. Ön konfigürasyon, X-ışını kristal yapısı gibi deneysel veriler kullanılarak üretilebileceği gibi yüksek çözünürlüklü spektroskopik yöntemlerden faydalınılarak da oluşturulabilir. Ön konfigürasyon olarak moleküler docking(kenetlenme) çalışmalarının sonuç çıktısından elde edilen veriler de olabilir.
Sistemin ön konfigürasyonu çözcü ile sarılır. Böylece ön konfigürasyondaki atomların birbiri ile olan etkileşimlerine, atomların çözücü ile olan etkileşimleri de dahil edilmiş olur. Sistemin biyolojik temelli simülasyonlarının en doğru sonucu vermesi için hesaplamaya özgü olarak çeşitli tuzlar var başka atomlar hesaplamaya dahil edilir.
Proteine kenetli ligand sisteminin MD simülasyonları için hazırlanması işlemine geçildi. Protein-ligand sistemi öncelikle “sistem oluşturucu” kullanılarak 10Å X 10Å X 10Å boyutlarında ortorombik hücre olacak şekilde OPLS3 kuvvet alanı kullanılarak çözücü ile sarıldı. Bu işlem GR’nin apo formu için aynı koşullarda tekrarlandı.
Yörünge Analizi(Trajectory) – Verilerin Yorumlanması
Kök Ortalama Kare Sapması (RMSD)
RMSD, x ve y’ye karşılık gelen atomlar arasındaki uzaklıkların karelerinin toplamının karekökü olarak ifade edilir. RMSD, iki durum arasındaki ortalama atomik yer değiştirmenin bir ölçüsüdür.
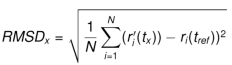
RMSD hesaplanırken tipik olarak ilk yapı referans olarak kullanılır. tref referans zamanı ve t = 0 olarak kabul edilir ve r ‘, x yapısındaki seçilen atomların, referans yapı üst üste bindirildikten sonra pozisyonudur, burada x durumu, tx zamanında kaydedilir. Bu işlem, simülasyon yörüngesindeki her durum için tekrarlanır.
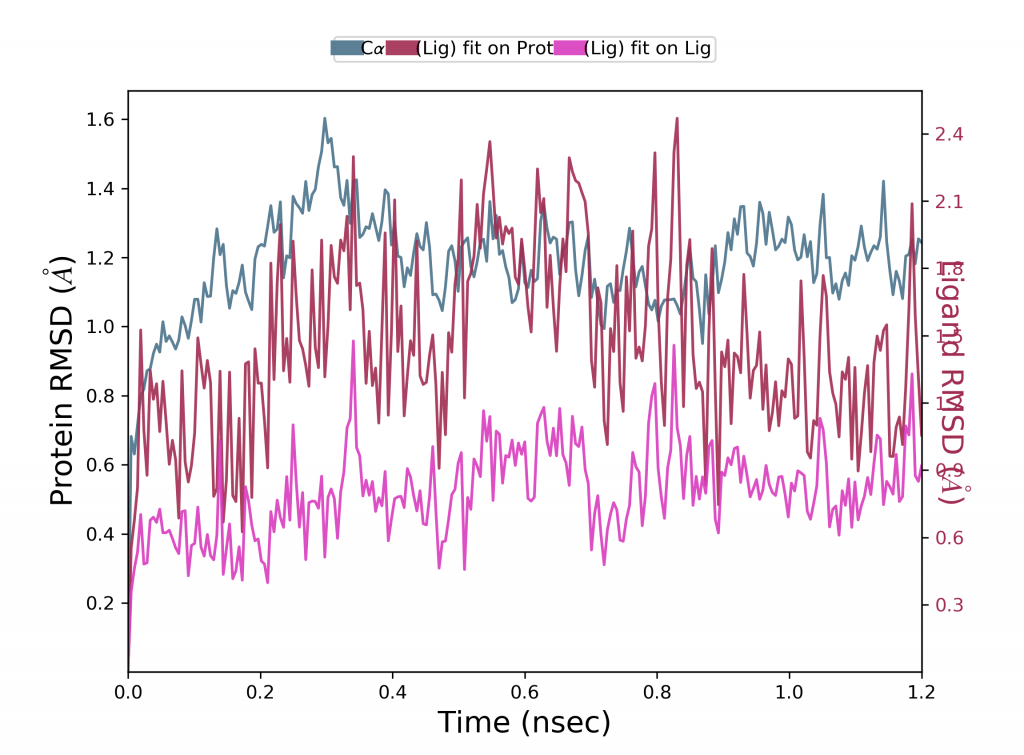
Protein RMSD
Lennard-Jones potansiyeli de burada hesaba katılmaktadır. Proteinin tüm yapısı önce referans yapı omurgası üzerinde hizalanır ve ardından RMSD, atom seçimine göre hesaplanır. Proteinin RMSD’sini izlemek, simülasyon boyunca yapısal konformasyonuna ilişkin içgörü sağlar. RMSD analizi, simülasyonun dengelenip dengelenmediğini gösterir. 1-3 Å sırasındaki değişiklikler, küçük, küresel proteinler için mükemmel şekilde kabul edilir. Bununla birlikte, bundan çok daha büyük değişiklikler, proteinin simülasyon sırasında büyük bir konformasyonel değişikliğe uğradığını gösterir. Simülasyonun sonunda proteinin RMSD’si ortalama olarak hala artıyor veya azalıyorsa, sistem dengelenmemiştir ve simülasyonunuz titiz analiz için yeterince uzun değildir.
Ligand RMSD
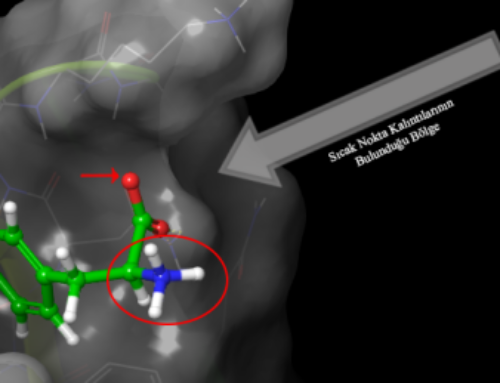
Lennard-Jones potansiyeli hesaplaması burada başta iki hidrojen atomunun birbirine göre konumlarına bakarken büyük önem kazanmaktadır. Ligand RMSD, ligandın proteine ve bunun bağlanma cebine göre ne kadar stabil olduğunu gösterir. Protein-ligand kompleksi ilk önce referansın protein omurgası üzerinde hizalandığında ve ardından ligand ağır atomlarının RMSD’si ölçüldüğünde bir ligandın RMSD’sini gösterir. Gözlenen değerler protein RMSD’den önemli ölçüde daha büyük olduğunda, ligand başlangıçtaki bağlanma bölgesinden uzaklaşmıştır.
Kök Ortalama Kare Dalgalanması (RMSF)
Lennard-Jones potansiyeli bu hesaplama formülüne bir şekilde dahil edilmekte ve yazılımsal olarak mümkün olduğunca doğru hesaplamalar yapılmaya çalışılmaktadır. RMSF, protein zinciri boyunca değişiklikleri analiz etmek için kullanılır. Aminoasit kalıntısı i için RMSF:
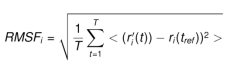
Olarak verilir. Burada T, RMSF’nin hesaplandığı yörünge zamanıdır, tref referans zamanıdır ve ri, i kalıntısının pozisyonudur; r ‘, referans üzerindeki üst üste bindirmeden sonra kalıntı atomlarının pozisyonudur ve köşeli parantezler, kalıntıdaki atomların seçiminde keresi alının mesafenin ortalamasının alındığını gösterir. Bu veride zirveler, simülasyon sırasında en çok dalgalanan protein alanlarını gösterir. Tipik olarak kuyrukların (N- ve C-terminalleri) proteinin diğer herhangi bölümlerinden daha fazla dalgalandığını gözlenir. Alfa sarmalları ve beta sarmalları gibi ikincil yapı öğeleri genellikle proteinin yapılandırılmamış kısmından daha serttir ve bu nedenle döngü bölgelerinden daha az dalgalanır. Lennard-Jones potansiyeli bu sefer protein yapının kendi atomları başta olmak üzere etkileşimlerinin hesaplanmasında kullanılmaktadır.
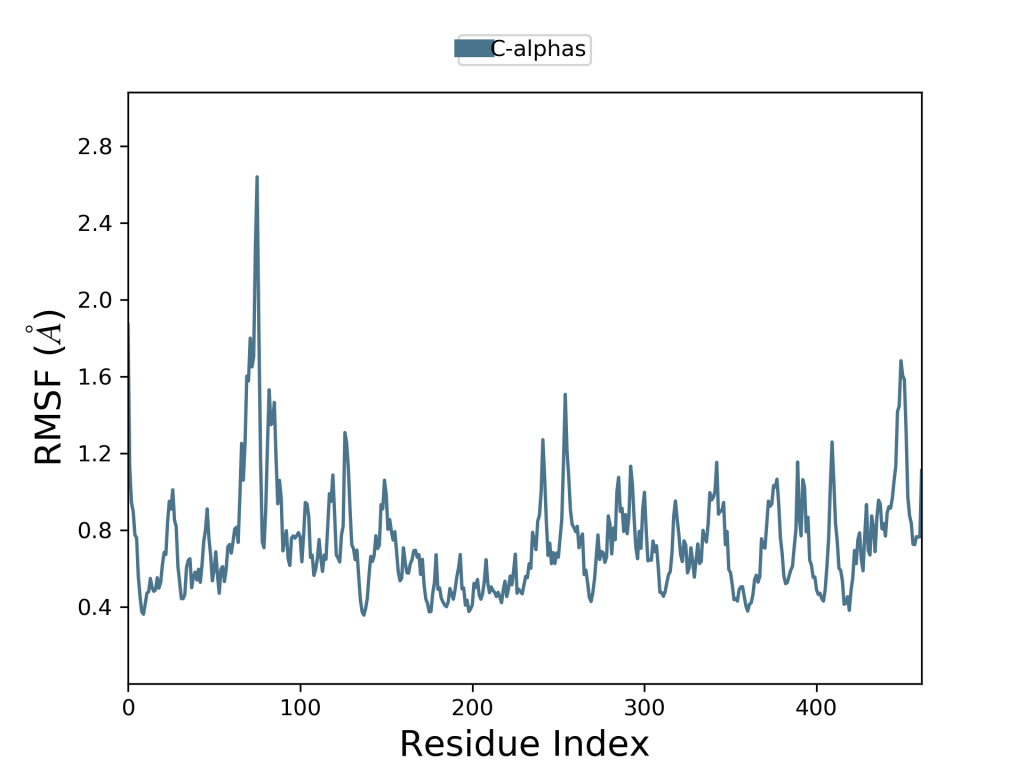
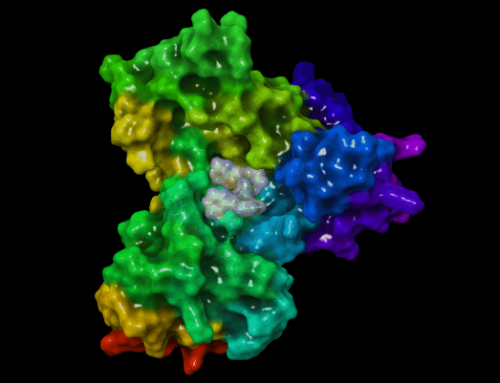
Burada Lennard-Jones potansiyeli ve yukarıda açıklanan daha pek çok yöntem ile oluşturulan hesaplama çıktısı görülmektedir. Bu çıktı bir protein yapının yakınında bir ligand varken nasıl hareket ettiğini göstermektedir. Hareket liganda özgüdür. Yani farklı bir ligand varlığında farklı bir hareket söz konusu olmaktadır. Ligand ve protein yapının bir arada olduğu baika bir örnek için Moleküler Dinamik Simülasyonları yazısını okuyabilirsiniz. Burada Glutatyon Redüktaz enzimine yapılmış bir docking çalışması bulunmaktadır. Glutatyon redüktaz enzim aktivitesi hakkında daha fazla bilgi almak için Glutatyon Redüktaz Enzim Aktivitesi adlı yazıyı okuyabilir ve Yanaştırma Nedir? Moleküler Docking Nasıl Yapılır? adlı yazıdan bu enzime nasıl yanaştırma(kenetlenme veya docking) yapıldığını öğrenebilirsiniz.