Antimalaryal İlaç Keşfi
Glutatyon redüktaz enzimi, canlı yapısında parazit organizmaların hayatta kalması için gerekli enzimlerden birisidir. Bu çalışmada oksitlenmiş glutatyonun, glutatyona dönüşümünü katalizleyerek parazit organizmanın hayatta kalmasını sağlayan glutatyon redüktaz enziminin inhibitör ligand ile engellemesi ile tedavi edici etkisi olan ilaçların keşfi hedeflenmiştir. Glutatyon redüktazın mormal zamanlarda, yani parazit organizmanııın metabolizmada faaliyet göstemediği zamanlarda antioksidan işlevinin hızlandırılması için de hesaplamalı kimya çalışmaları yapılmıştır. Yani bu çalışmanın ana amacı antimalaryal ilaç keşfi ve normal zamanlarda da antioksidan işlevinin hızlandırılması için aktivatör molekül keşfidir. Bu kapsamda bu ligandların keşfi için yapı bazlı ilaç keşfi bilgisayar destekli ilaç tasarımı araçlarından olan moleküler docking ve moleküler dinamik çalışmaları yapılmıştır.
Antimalaryal İlaç Tasarımında Sanal Tarama
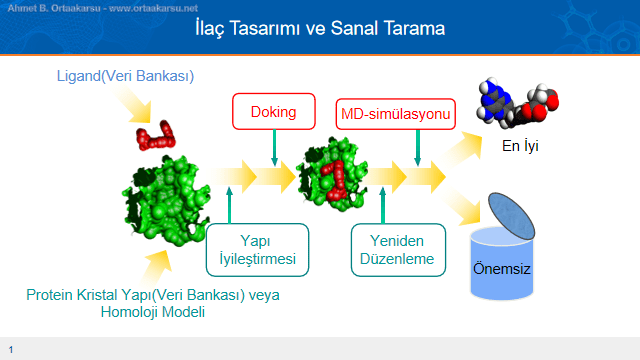
Yapı bazlı ilaç tasarımında ligandlar ve protein yapı veri bankalarından seçilir. Sonrasında bu yapılar belirli kriterlerle hazırlanır. Ligandlar veri bankalarından belirli fonksiyonel gruplara sahip olacak şekilde seçilebileceği gibi(yani antimalaryal benzeri ligandlar, herbisit benzeri ligandlar şeklinde) herhangi bir kriter gözetmeksizin de seçilebilir. Ligandların sayısı çoğu zaman yüzbinleri bulabilmektedir. Bu ligandlar için çalışılacak pH’da molekül içi düzenlenme belirli yazılımlarla tahmin edilir ve ligandlar ilgili pH’da moleküler docking işlemi için hazır hale getirilir.
Protein yapı ise belirli bir hedef doğrultusunda seçilir. Eğer hedef protein yapının PDB’de verisi yok ise homoloji modelleme ile hedef yapının tahminsel yapısı hazırlanabilir. Bunun sonucunda protein yapıdaki eksik kalan amino asitler tamamlanıp çalışmanın yürütüldüğü ortamın pH değeri ayarlanır, yapıdaki hidrojen bağları bu pH için modifiye edilir ve minimize etme işlemi sonucunda protein yapı moleküler docking işlemine hazır hale getirilir. Bu, hazır hale getirme işlemine “yapı iyileştirmesi” denilmektedir.
Moleküler docking işlemininden önce belirli ayarlamalar ile protein yapıda doking(kenetlenme) protokollerinin uygulanacağı bölgeler yazılıma bildirilmelidir. Bunun nasıl yapıldığını görmek için Yanaştırma Nedir? Moleküler Docking Nasıl Yapılır? yazısını okuyabilirsiniz.
Akabinde gerçekleştirilen moleküler docking(yanaştırma veya kenetlenme) işlemi ile en iyi kenetlenme özelliklerine sahip ligandlar belirli skor algoritmaları ile belirlenir ve en yüksek puanlı olan ligandlar için moleküler dinamik çalışmalarının hazırlıklarına başlanır. Bu çerçevede tüm yapı(protein yapı ve ona kenetli ligand) moleküler dinamik çalışmalarının gerçeği yansıtması için metabolizma ile aynı çözücü ortamına konulur. Bu yapının hazırlanması için “yeniden düzenlenme” adımıdır.
Moleküler dinamik hesaplamalarının nasıl çalıştığını anlamak için Moleküler Dinamik Simülasyonları ve Lennard Jones Potansiyeli ve Moleküler Dinamik Simülasyonlarında Kullanımı adlı yazıları okuyabilirsiniz.
Moleküler dinamik çalışmaları ile docking(kenetlenme) işlemi sonucunda iyi kenetlenme sonuçları elde etmiş ligandlar arasından da eleme yapılır. Yani protein yapıya en iyi kenetleme özelliği gösteren, protein yapının normal hareketini en iyi engelleyen ligand moleküler dinamik verilerine bakılarak karar verilir.
Antimalaryal Aktivite ve Moleküler Docking
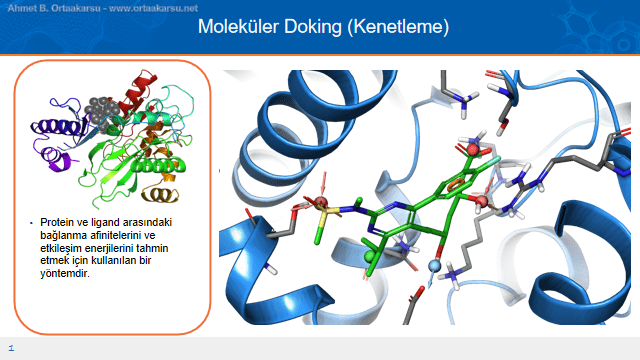
Moleküler docking, ligand ve protein arasında bağlanma afinitelerini tahmin etmeye dayalı bir yöntemdir. Moleküler docking prosedürünün nasıl işlediğini ve kullanım alanlarını öğrenmek için Moleküler Docking ve İlaç Tasarımında Kullanımı adlı yazıyı okuyabilirsiniz. Moleküler docking günümüzde favicovir ve diğer Covid-19 ilaçlarının keşfinde kullanılmaktadır. Favicovir ve Koronavirüs İlaç Geliştirme Çalışmaları adlı yazılarda bu konularda çokça bahsedilmektedir. Favipiravir, remdesivir ve plaquenil diğer ilaçlardır. Favipiravir Nedir? Favipiravir Yan Etkileri Nelerdir? ve Plaquenil Yan Etkileri Nelerdir? adlı yazılarda bu ilaçlardan ayrıntısıyla bahsedilmektedir.
Moleküler Docing Nedir? Nasıl Çalışır?
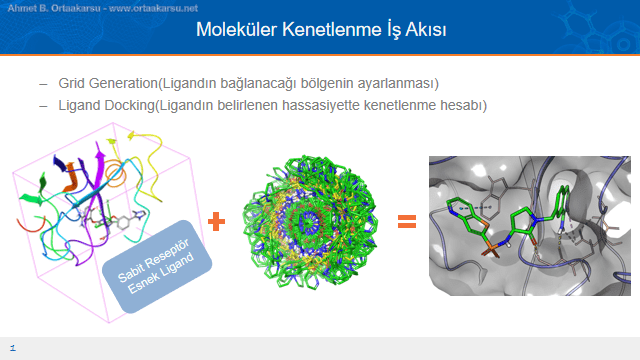
Moleküler kenetlenme, yani moleküler docking; özetlersek, protein yapıda kenetlenilecek yerin gösterilmesi sonucu bu yere ligandların olası tüm konformasyonlarının denenmesi ile en iyi bağlanma modlarının tahmin edilmesi üzerine kurulu bir sistemdir. İyi bağlanan bir konformasyonun “ıskalanmaması” için birden fazla sayıda konformasyon her bir ligand için ürerilir ve her bir konformasyon protein yapıda kenetlenme için gösterilen yere denenir.
Moleküler Docking ve Doğrulama
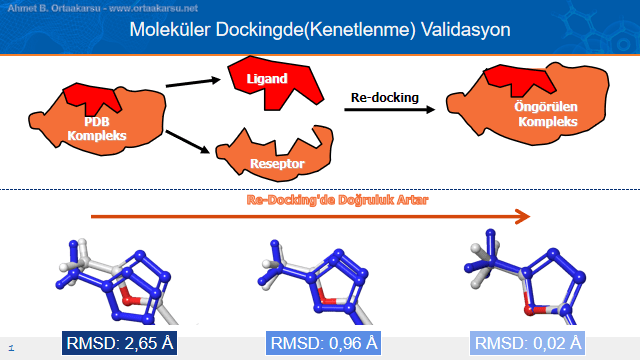
Moleküler docking çalışması yaparken (buna antimalaryal ilaç keşfi, genel itibari ile kanser ilaçlarının keşfi, Raltegravir – İntegraz İnhibitörlerinin İlki ve bunun gibi pek çok çalışma da dahildir) genel olarak kristal verisi bulunan protein yapının içerisindeki ligandın çıkarılıp tekrar kenetlenmesi ile doğrulanır. Bunda amaç deneysel olarak elde edilen kristal verisindeki ligandı, yine kristal yapının içerisine aynı şekilde kenetlemektir. Kristal yapının içerisine, kristal yapıdan çıkarılmadan önceki gibi kenetlenen ligandın, kenetlenme ayarları kullanılarak diğer ligandlar için de gerçeğe en yakın kenetlenme protokolleri ve ayarları kullanılır. Bunun anlamı deneysel sonuca yakın veri anlamına gelmektedir. Ancak bunun, kristal yapının içerisindeki liganda benzeyen ligandlar söz konusu olduğunda yüzde yüz sonuç verdiği, benzer olmayan ligandlarda hatalar olabileceği unutulmamalıdır.
Yukarıdaki şekillerden alt tarafta olanlar RMSD olarak ifade edilen sapma değeridir. Bu değerin büyüklüğü aynen şekilde görüldüğü gibi ligandın tekrar kenetlenmesinde ne kadar başarılı olduğunu göstermektedir. Değer ne kadar büyürse tekrar docking işleminde o kadar fazla sapma olduğunu gösterir. Son şekil ve RMSD değeri görüldüğü gibi çok ideal bir çalışmanın ürünüdür. Bu da bu liganda benzer ligandlar bu yapıya kenetlendiği sürece oldukça doğru sonuçlar alınacağı anlamına gelir. Antimalaryal ilaçların keşif çalışmaları oksitlenmiş glutatyona benzer ligandlar kullanılarak gerçekleştirilmiştir. Bu tekrar docking sonucu elde edilen RMSD değeri son derece küçüktür. Bu yüzden verilerin son derece ideal olduğu söylenebilir.
Antimalaryal Aktiviteyi Anlatmadan Önce Moleküler Dinamik
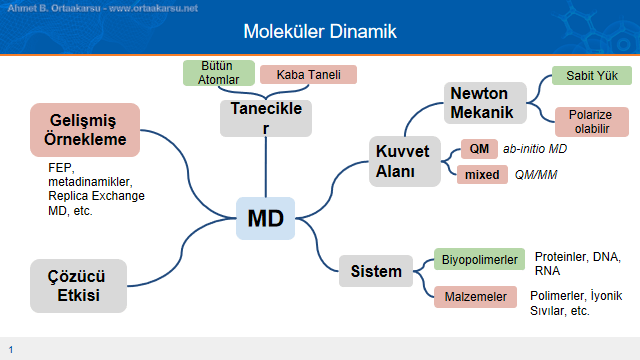
Moleküler dinamik çalışmalarının kullanım alanları sadece antimalaryal ilaçlar veya protein ligand etkileşiminin hesaplanması ile sınırlı değildir. Moleküler dinamik hesaplamaları malzeme biliminden polimerlere kadar pek çok alanda kullanılmaktadır. Bunların arasında DNA hesaplamaları, yüzey absorplama, metal yüzey etkileşimlerinin modellenmesi, protein ligand etkileşimi, çeşitli biyolojik mutasyonların öngörülmesi gibi çok çeşitli konular vardır. Burada temel ayrım moleküler dinamik çalışmalarında kullanılan hesaplamalarla ilgilidir.
Protein-ligand etkileşiminde çözücü etkileşimi yoğun olarak hesaba katılırken, metal yüzey modellemesinin yapıldığı(örneğin alüminyum yüzeyin suyu absorplama kabileyitinin incelenmesi) hesaplamalarda çözücü etkisi gibi bir kavram kullanılmamaktadır. Bir başka örnek olarak anti body ilaç keşfinde yoğun olarak bağ yapmayan etkileşimlerin hesaplamaya dahil edilmesi gösterilebilir.
Moleküler dinamikte, modellemenin yapılacağı sisteme göre kuantum mekaniği veya moleküler mekanik seçilebilir ve hesaplama istenen şekilde yaptırılabilir. Burada özellikle çok atomlu sistemlerde kuantum mekaniğinin kullanılması hesaplama süresinin uzunluğu açısından sorun teşkil edebilir. Antimalaryal ilaç keşif çalışmalarında olduğu gibi moleküler mekaniğin kullanılması hesaplamanın doğruluğunu bir miktar etkilese de deneysel sonuçlar(in vitro) son derece gerçeğe yakın olduğunu göstermiştir. Kuantum mekaniği ise az atomlu sistemlerde kullanılır. Sonuçlar ise genel itibari ile oldukça doğru olmaktadır.
Moleküler Dinamik Nedir?

Moleküler dinamik simülasyonları protein-ligand etkileşimi başta olmak üzere son derece sık kullanılan yöntemlerdir. Özetle parçacıkların birbiri ile olan etkileşimlerinin modellenmesi ve simülasyon haline getirilmesidir. Burada kuvvet alanı kavramı oldukça önemlidir. Çünkü çalışılan sisteme göre uygun kuvvet alanının seçilmesi veya seçilememesi sonuçların doğruluğunu büyük ölçüde etkilemektedir. Biz protein-ligand etkileşiminde oldukça sık kullanılan ve yerinde hesaplama kriterleri içeren OPLS_2005 kuvvet alnını kullanarak antimalaryal ilaç keşif çalışmalarını gerçekleştirdir. Moleküler docking ve moleküler dinamikte farklı kuvvet alanlarını tercih ettik. Bu konu ile daha fazla bilgi için Moleküler Dinamik Simülasyonları adlı yazıyı okuyunuz.
Moleküler Dinamik ve Kuvvet Alanları
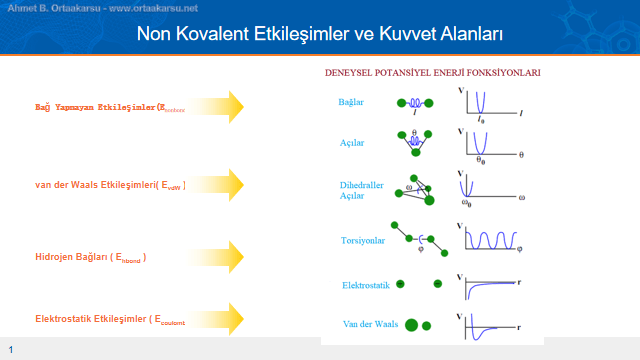
Kuvvet alanları hesaplamaya kattıkları etkileşim türleri bakımından farklılık göstermektedir. Kovalent olmayan etkileşimlerden iki atom alasında kovalent etkileşimlere, burulma ve torsiyon açılarındaki değişikliklere kadar pek çok etkileşim türü kuvvet alanlarının konusunu oluşturmaktadır. Kuvvet alanlarının hesaplamaya kattığı etkileşimlerin hesaplanma şekilleri kuvvet alanına göre değişiklik göstermektedir ancak hepsi matematiksel bir fonksiyon olarak ifade edilir. Bu etkileşim türleri bağ yapmayan etkileşimler, van der Waals etkileşimleri, hidrojen bağları(özellikle protein-ligand etkileşiminde önemli bir yeri vardır), elektrostatik etkileşimler hesaplamaya katılan temel etkileşimlerdir.
Moleküler Docking ve Moleküler Dinamik
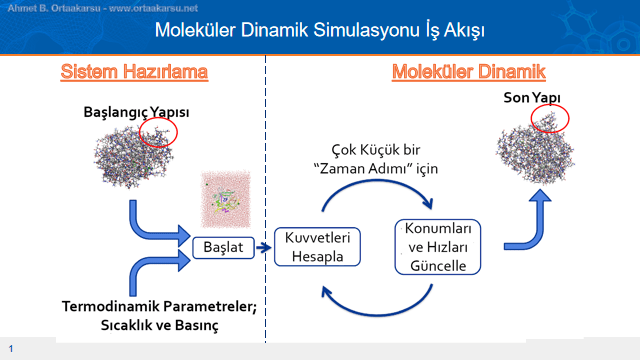
Yapı bazlı ilaç tasarım ve keşif çalışmalarında moleküler dockingden moleküler dinamiğe uzanan bir iş akışı söz konusudur. Burada amaç yazının ilk kısmında olduğu anlatıldığı gibi etkili bir ligand elemesi yapmaktır. İki yüz bin tane ligandın bir veya bir kaç liganda indirilmesi için son adım en başarılı şekilde kenetlenen ligandların protein yapıya kenetli şekilde doğal ortamında modellenmesi işlemidir. Bunun için yüksek sayıdaki ligandları önce yüksek hızlı sanal tarama, sonrasında standart hassasiyetli tarama ve en sonunda da yüksek hassasiyetli sanal tarama şeklinde, adım adım Maestro Yazılımını kullanarak eliyoruz ve elde edilen bir kaç ligand moleküler dinamik simülasyonları ile protein yapıya doğal ortamında ne kadar süre tutunuyor ve inhibisyon işlemi gerçekleştiriyor bakıyoruz.
Yukarıdaki şekilde bu sürecin moleküler dinamik simülastonları ile ilgili olan adımının nasıl işlediği anlatılmaktadır. Burada protein-ligand kompleksi önce belirli parametreler girilerek hazırlanır. Bu parametrelerin başında sıcaklık ve basınç gelmektedir. Sonrasında, simülasyon işlemini başlatmadan hemen önce protein-ligand kompleksi protein yapının doğal ortamına en yakın şekilde hazırlanır. Bu aşamada protein yapı çözücü ile sarılır. Çözücü, protein yapının doğal ortamındaki ile aynı olmalıdır. Eğer protein yapının doğal ortamında başka iyonlar varsa bu aşamada dahil edilir.
Antimalaryal ilaç keşif çalışmamızda bu ortam glutatyon redüktaz enziminin doğal ortamı olan sudur. Kanın ve insanın çok büyük bir bölümünün sudan oluştuğu düşünüldüğünde protein yapının su ile sarılması gereklidir. Moleküler dinamik çalışmalarına bu aşamada, metabolizmadaki işleyiş ve protein yapının yeri bakımından çeşitli iyonlar eklenmesi gerekebilir.
Sonrasında moleküler dinamik simülasyonun başlaması ile her zaman adımında atomlar arası etkileşimler kuvvet alanına göre hesaplanır ve belirlenen sürenin sonucunda simülasyon son bulur. Protein-ligand komplekisinin çözücü etkileşimi sonunda hem ligandın hem de protein yapının(şekilde işaretlendiği gibi) konformasyonel değişimler geçirdiği görülebilir.
Glutatyon Redüktaz Enzimi İçin Yapı Bazlı İlaç Tasarım Çalışmaları

Glutatyon redüktaz enziminin önemini ve parazitlerdeki yaşam sürecinin bir parçası olduğunu daha önce söylemiştim. Glutatyon redüktaz enzimi sağlıklı bir metabolizmada önemli görevleri olan bir enzimdir. Bu çerçevede, parazit organizmanın bulaşması sonucu inhibe edilmesi, parazit organizma yokken ise gerek duyulduğunda aktive edilerek etkinliğinin arttırılması önem taşımaktadır. Bu çerçevede yeni sentezlenen Schiif Bazlarımızın bu enzim üzerindeki etkisini yapı bazlı ilaç tasarım yöntemleri kullanarak belirledik.
1GRA Kodlu Glutatyon Redüktaz
PDB: 1GRA(DOI: 10.2210/pdb1GRA/pdb) olan protein yapıyı çalışmalarımızda kullandık.
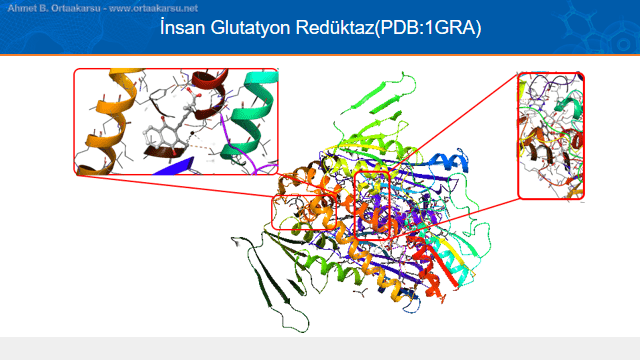
Glutatyon redüktaz enzimi içerisinde enzime sıkı sıkı bağlı FAD kofaktörü ve başka bir bölgesine bağlı NAD ile şekildeki gibi görülmekte. Bu yapıda ayrıca şekilde gösterilmesi mümkün olmayan GSSG molekülü aktif merkezin yakınında bulunmakta. Moleküler docking ve moleküler dinamik çalışmalarının yapılacağı protein yapıda daha öncesinden bahsedilen hazırlık evresinden geçildi. Eksik her hangi bir amino asit kalıntısı olmadığı belirlendi ve protein yapı ileride kullanılmak üzere hazırlandı.
Çalışmada Kullanılan Schiff Bazları
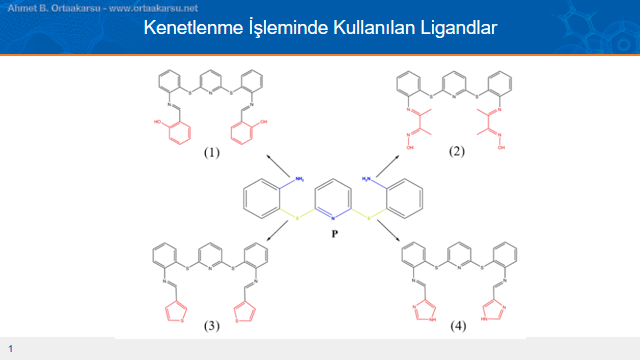
Yukarıdaki Schiff bazları öncelikle moleküler docking hesaplamaları için daha önce bahsedilen şekilde hazırlandı.. Bu Schiff bazları şekilde orta kısımda görülen ve P ile ifade edilen yapının türevleri şeklinde sentezlenmiştir. P apısıda dahil tüm türevler moleküler docking çalışmalarında kullanılmış ve hesapmala sonuçları değerlendirilmiştir.
Antimalaryal Aktivite İçin Moleküler Docking Sonuçları
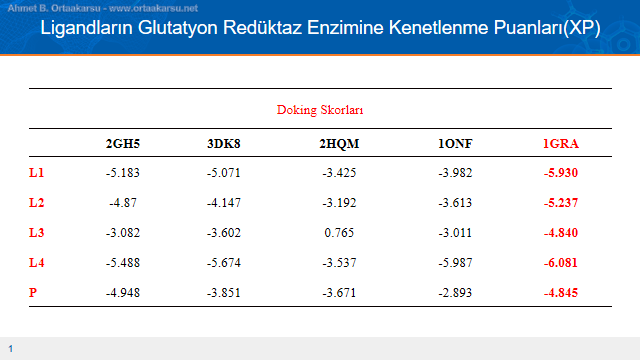
Moleküler docking hesaplamalı sonucunda yukarıdaki tablo elde edilmiştir. 1GRA yapısının dışında başka PDB kodlu protein yapılar için de moleküler docking işleminin sonuçları görülmektedir. Bu sonuçlar daha önce yapılan çalışmaların sonuçlarıdır. Görüldüğü gibi 1GRA için yapılan moleküler docking hesaplaması oran olarak diğer protein yapılara yapılan moleküler docking çalışmaları ile uyum içindedir. Her protein yapıda aynı sıra ile puanlama karşımıza çıkmaktadır.
Tabloda görülen ve en yüksek doking skoruna sahip olan L4 ligandı moleküler docking çalışmalarından sonra en iyi kenetlenen ligand olarak moleküler dinamik simülasyonuna önceden belirtilen hazırlık adımları sonucunda tabi tutulmuş ve belirli sonuçlar alınmıştır. Ancak bu kısma geçmeden önce 1GRA kodlu protein yapıya kenetli ligandların en yüksek skora sahip olanlarının analizi yapılacaktır.
L1 Ligandının Glutatyon Redüktaz Enzimine Kenetlenmesi
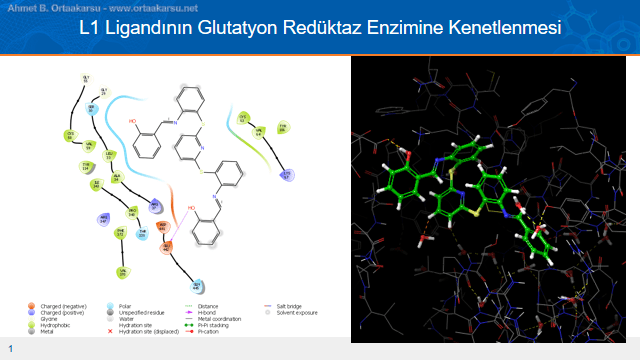
Docking skorlarını içeren tabloya baktığımızda L1 ligandının oldukça yüksek bir skora sahip olduğu görülmektedir. L1 ligandı, L4 ligandından sonra protein yapıya en iyi kenetlenen ligand olarak karşımıza çıkmaktadır. Yapının hem iki boyutlu hem de üç boyutlu şekillerinde görülen -OH’da hidrojen protein yapıda gulutamin(442. aminoasit) kalıntısına kuvvetli hidrojen bağı yapmaktadır. Molekülün şeklinin de protein yapıya oldukça iyi uyum sağladığı görülmektedir. Ligandın bir bölümünün de protein yapının iki boyutlu görüntüsünde kalın mavi çizgi ile gösterilen “oyuk” kısma oldukça uyumlu ve iyi yerleştiği açıktır. Yüksek docking skoru ve iyi yerleşmiş şekli ile L1 ligandı glutatyon reduktaz enzimi için iyi bir inhibitör adayıdır. Antimalaryal ilaç adayı olarak daha gelişmiş yöntemlerle(moleküler dinamik) analizi yapılabilir. Bu ligandın in vitro çalışmaları ile ilgili verilere ulaşmak için Glutatyon Redüktaz Enzim Aktivitesi yazısını okuyabilirsiniz. İn vitro çalışmalar göstermektedir ki bu ligand glutatyon reduktaz enzimini inhibe etmektedir.

L4 Ligandının Glutatyon Redüktaz Enzimine Kenetlenmesi
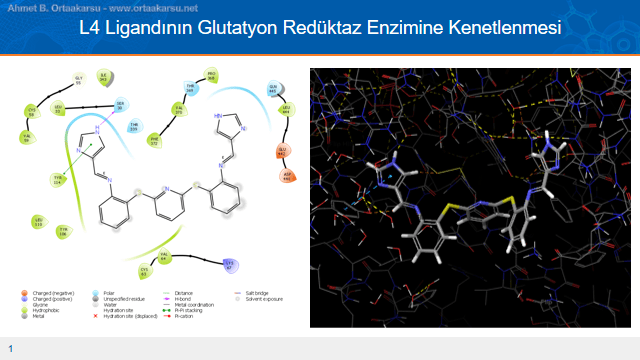
Docking skorlarının olduğu tabloda en yüksek puana sahip olan L4 ligandıdır. L4 ligandı oldukça aktif olan imidazol halkası içermektedir ve bu özelliği ile diğer ligandlardan ayrılır. L4 ligandının imidazol halkasında -NH’ın hidrojeni serin(30. aminoasit) kalıntısı ile hidrojen bağı yaparken, yine aynı imidazol halkasında tayrosin(114. aminoasit) kalıntısı ile pi-katyon etkileşimleri görülmektedir. Dolayısıyla ligandımızın bu kısmı protein yapıya oldukça iyi bir şekilde kenetlenmiştir.
Bu etkileşimler ve molekül yapısının protein yapıdaki “oyuk” kısımlara oldukça uyumlu yerleşmesi L4 ligandına en yüksek docking skorunu sağlamıştır. Görsellere baktığımızda hem iki boyutlu resimde hem de üç boyutlu resimde etkileşimler açıkça görülmektedir. L4 ligandının protein yapıya kenetlenmesinin oldukça simetriktir. Ligandın iki ucunda bulunan imodazol halkalarının “oyuklara”(veya “ceplere”) girdiği görülmektedir.

Yukarıdaki görsel ligandımızın protein yapıdaki ceplere oldukça iyi uyum sağladığını göstermektedir.
Antimalaryal İlaç Tasarıında Moleküler Dinamik
Antimalaryal ilaç olarak etkin molekülün bulunması için docking skorları ile yapılan değerlendirme sonucunda en yüksek puana sahip olan ligandlar seçilerek daha ileri hesaplama teknikleri ile sonuçlar analiz edilmelidir. Sadece moleküler docking çalışmaları büyük ölçüde doğru sonuç verse de kesin sonuçların tespiti moleküler dinamik hesaplamaları ile yapılmaktadır. Moleküler dinamik çalışmaları için daha önceden ayrıntılı şekilde anlatılar hazırlık evrelerine geçilir.
Moleküler Dinamik Sisteminin Hazırlanması
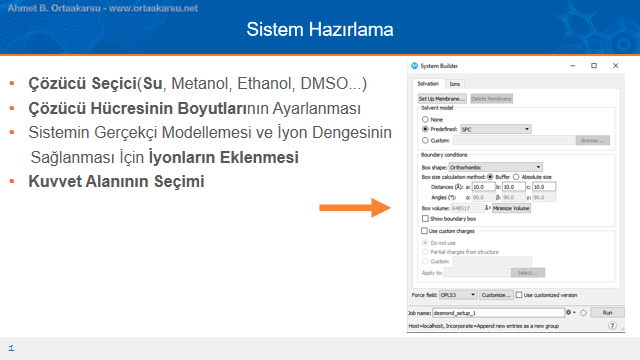
Antimalaryal ilaç keşif çalışmalarında ligandlar arasında yapılan eleme işleminin hesaplamalı kimya adına son adımlarından birisi moleküler dinamik simülasyonlarıdır. Bu hesaplama ile ligandımızın glutatyon redüktaz enzimini inhibe etme potansiyeli belirlenecektir. Bunun için ligandın protein yapıda uzun süre kalması ve hesaplama süresi boyunca protein yapının alfa karbon omurgasının hareketinin sınırlanması gibi etkiler göz önünde bulundurulur.
Moleküler dinamik çalışmalarında, sistemin hazırlık evresinde yukarıdaki görsel ile açıklanan pek çok opsiyon seçilebilir. Yapılan seçimler tamamen ilgili protein yapının doğal ortamındaki gibi veya doğal ortamına en yakın şekilde olmalıdır. Burada çözücü seçimi(biyolojik sistemler için genellikle su) yapılır ve çözücü moleküllerinin protein yapıyı kapladığı alanın şekli belirtilir. Ortorombik ve bunun gibi. , Burada antimalaryal ilaç adaylarının bulunması ile ilgilendiğimizden çözücü su olarak seçilmiştir. Çözücüyü oluşturacak olan hücre ise ortorombik seçilmiştir. Diğer ayarlar “varsayılan” şeklinde bırakılmıştır.
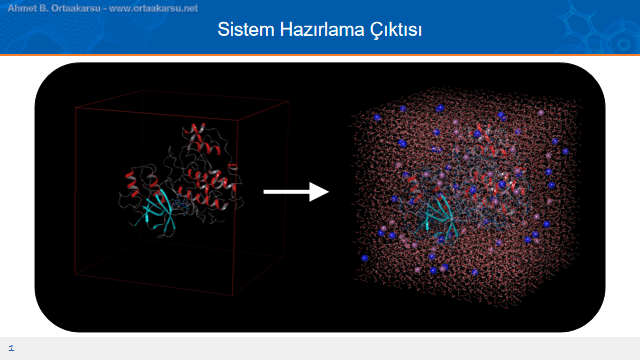
Sistem hazırlama çalıştırıldıktan sonra yukarıda görüldüğü gibi protein-ligand kompleksi çözücü molekülleri ile belirlenen hücre şekli çerçevesinde sarılmış durumda. Artık moleküler dinamik simülasyonu çalıştırılarak atomların birbiri ile olan etkileşimleri zamana bağlı olarak izlenebilir.
Antimalaryal aktivite için Moleküler Dinamik Simülasyonu Çalıştırılıyor
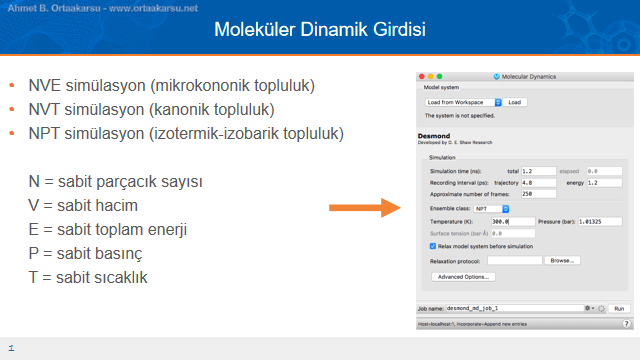
Antimalaryal aktiviteyi hesaplamalı kimya ile denemek için moleküler docking yapıldı ve artık moleküler dinamik simülasyonunun çalıştırılma aşamasına gelindi. Simülasyonu çalıştırmadan önce çalışma alanındaki protein-ligand kompleksi ve çözücülerin hepsini “load” ediyoruz. Yani hesaplama yapılacak sistem olarak gösteriyoruz. Sonrasında hesaplama süresi, basınç ve sıcaklık gibi opsiyonları giriyoruz ve antimalaryal etkisinin docking skorları ile yüksek olacağı sonucuna vardığımız ligandın moleküler dinamik simülasyonu başlatılıyor.
Hesaplamanın ne zaman biteceği bilgisayarımızın “gücü” ile ilgilidir. Özellikle iyi bir grafik işlemcisine sahip bilgisayarlar simülasyonları kısa sürede tamamlayabilmektedir.
Antimalaryal İlaç Aktivitesi ve Moleküler Dinamik Verilerinin Yorumlanması
Moleküler Dinamik Simülasyonları yazısında anlatıldığı gibi enzime hem L4 ligandı bağlı iken hem de her hangi bir ligand bağlı değil iken moleküler dinamik simülasyonları yapılmıştır. Bundan sonra yapılacak analizlerin anlaşılması için Moleküler Dinamik Simülasyonları adlı yazıda anlatılanların bilinmesi önemlidir. Burada amaç ligand bağlı halin hareketinin enzimin normal hareketine engel olup olmadığını belirleyebilmektir. İlgili yazıda yorumlamanın nasıl yapıldığı ve verilerin değerlendirilmesi ayrıntısıyla anlatılmaktadır. Şimdi moleküler dinamik simülasyonları yapılan ligandların antimalaryal aktivite gösterip göstermediği belirlenecektir.
RMSD Grafiği
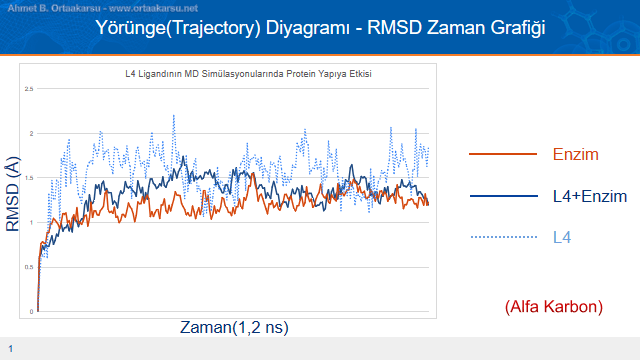
Kesikli çizgi ile gösterilen ligandın hareketidir. Koyu mavi çizgi ise ligand bağlı iken enzimin hareketidir. Bu iki verinin zamana bağlı olarak değişimi L4 ligandının enzime kenetli kalma potansiyelin bir göstergesidir. Görülmektedir ki ligandın hareketi, L4 ligandı kenetli ligandın hareketinden daha fazla sapma göstermektedir. Bu da L4 ligandının, docking puanı yüksek olsa da enzimin bu bölgesine uzun süre bağlı kalamayacağını göstermektedir. Protein yapının alfa karbonlarının zamana göre RMSD değerini incelediğimizde; apo formun hareketinin, halo formun hareketinden daha küçük RMSD değerine sahip olduğu görülmektedir. Yani L4 ligandı enzime bağlandığında enzimin hareketi sınırlanmamış, tam aksine artmıştır. L4 ligandının inhibisyon yapmadığı oldukça açıktır. Bu veriler RMSF grafiği ile desteklenerek daha net bilgiler elde edilecektir.
RMSF Grafiği
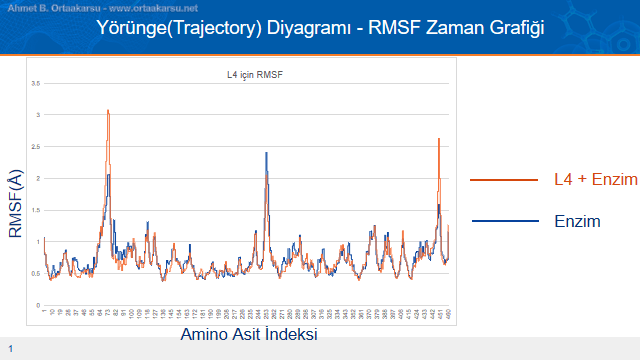
Yapı bazlı ilaç tasarımı çalışmalarında ikinci analiz yöntemi RMSF grafikleridir. Bu grafikte turuncu olan enzimin ligand bağlı olmadan ki verisi koyu mavi olan ise L4 ligandı bağlı olan verisidir. Bu veriler enzimin ligand bağlı halinin pek çok bölgesinde hareketin sınırlanmadığını açıkça göstermektedir. Özellikle baştaki ve sondaki göze çarpan pikler bu durumun açık birer ifadesidir. İnhibisyon olan herhangi bir durumda L4 + Enzim verileri, sadece enzimin verileri ile karşılaştırıldığında, enzimin verilerinin altında kalması gereklidir. Bu RMSF grafinde ise bu beklenti karşılanamamıştır.
Protein-Ligand Etkileşim Analizi
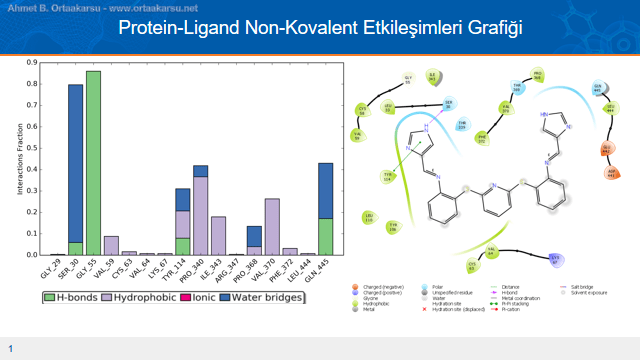
Yukarıdaki grafikte sağ tarafta L4 ligandının protein yapıya kenetli halinin iki boyutlu hali gösterilmekte, sol tarafta ise protein-ligand etkileşimlerinin zamana bağlı olarak nasıl değiştiği görülmektedir.
Moleküler dinamik simülasyonu başlatıldığı andan itibaren iki boyutlu yapıda arka tarafta görülen gilisin(55. amino asit) kalıntısı ile simülasyon süresi boyunca yoğun bir bir etkileşim başladığı görülmektedir. Burada söz konusu olan oldukça güçlü hidrojen bağıdır. Ayrıca su köprüsü oluştuğu görülmektedir. Tüm bu etkileşimler moleküler dinamik simülasyonu boyunca molekülün bu etkileşimleri yapan bu kısmının, protein yapıya bağlı kaldığı görülmektedir. Simülasyonun başladığı ve bittiği yapıya iki boyutlu baktığımızda bu durum daha anlaşılır olacaktır.
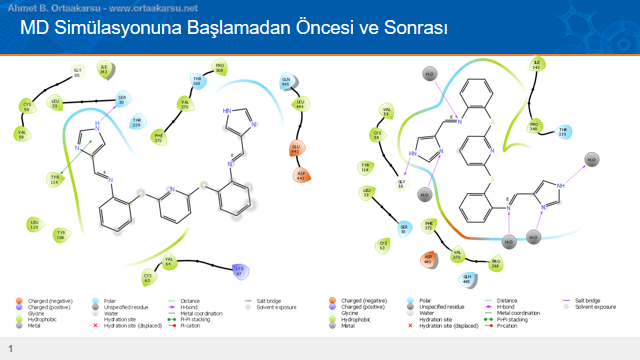
Gri dairelerle gösterilen su molekülleri çözücü ile sarılma işlemi gerçekleştirildiğinde eklenen çözücü molekülleridir. Bu çözücü molekülleri ligandın öbür ucu(glisin amino asiti ile etkileşmeyen taraf) ile yoğun etkileşimler oluşturmuştur ve schiff bazındaki azot ile etkileşim içerisine girerek molekülün konformasyonun değişimine katkı yapmıştır.
Glisin ile etkileşen kısımda ise pi-katyon etkileşimleri yerini hidrojen bağlarına bırakmıştır. Bu etkileşimlerden birisi azor ve su molekülü arasında kurulan hidrojen bağıdır. Başlangıç ve bitiş iki boyutlu görüntülerine baktığımızda konformasyonel değişimde çok açık bir şekilde görülmektedir. Konformasyonel değişim üç boyutlu görüntüde daha net görülmektedir.


Başlangıç ve bitiş görüntüleri karşılaştırıldığında hem protein yapıda hem de ligandda konformasyonel değişim açıkça görülmektedir. Görüntüde kırmızı bölgeler kısmi negatif bölgeler iken mavi bölgeler kısmi pozitif bölgelerdir. Mavi ve yeşil alanların başlangıç ve simülasyon sonu durumlarına baktığımızda protein yapıdaki değişim görülmektedir.
L4 ligandının gilisin(55. amino asit) kalıntısı ile etkileşen imidazol halkasının protein yapının içerisinde neredeyse hiç oynamadığı görülmektedir. İmidazol halkası buradaki “cebin” dışına hiç çıkmamakta, ligandın konformasyonel değişimi ise diğer uçtaki imidazol halkasından itibaren olmaktadır.
Sonuç
Antimalaryal ilaçların yapı bazlı ilaç tasarım çalışmaları ile keşfi için yapılan moleküler docking ve moleküler dinamik simülasyon çalışmaları sonucunda veriler elde edilmiş ve yorumlanmıştır. Oldukça yüksek docking puanı ile moleküler dinamik simülasyonları gerçekleştirilen L4 ligandı, docking skorunun aksine moleküler dinamik verilerine göre iyi bir inhibisyon yapmamaktadır. Bu sonuca başta alfa karbonların hareketinin sınırlanamaması ile varılmış, RMSF grafiği ile bu sonuç desteklenmiştir.
Glutatyon redüktaz enzimini aktive ettiği bilinen L4 ligandının moleküler dinamik verileri bu durumu destekler niteliktedir. L4 ligandının inhibisyon yaptığına dair hiç bir sonuç bulunamamıştır. L4 ligandı antimalaryal aktivite göstermemektedir. Tam aksine in vitro çalışmalarında ışığında bu ligandı glutatyon redüktaz enzimini aktive ettiği görülmüştür. L4 ligandı aktivatör bir bileşik olarak antioksidan eksikliğini giderme amacıyla kullanıma olasılığı olan bir bileşik olarak karşımıza çıkmaktadır.
Bu konuda yapılan çalışmaların yetersiz olması L4 ligandının bu amaçla kullanılabileceğine dair kesin veriler olmadığı anlamına gelmektedir. L4 ligandı istisnai bir bileşik olarak farklı araştırma konuları için kullanılabilir. Antimalaryal aktivite için kullanılma potansiyelinin olmadığı ise oldukça kesindir. Antimalaryal aktivite için kullanılma potansiyeli olan ligand ise docking skoru baz alındığında ikinci sırada olan L1 ligandıdır.
Bu çalışmada verileri paylaşılmayan L1 ligandı, L4 ligandının verilerinin tam aksine, ikinci sıradaki yüksek docking puanı ile iyi bir inhibitör olduğunu göstermektedir.