Yeni ilaçların geliştirilmesi için in siliko yöntemler giderek daha fazla kullanılmaktadır. Bilgisayar destekli ilaç tasarımı (CADD) ilaç-reseptör, ilaç-enzim etkileşimlerini simüle etmek için hesaplama yöntemlerini kullanan bir disiplindir. Kimyasal moleküllerin 3-D özelliklerini inceleyerek yapılan hesaplamalar öncü bileşiklerin optimizasyon sürecini hızlandırmaktadır. Böylece, ilaç araştırma ve geliştirme (Ar-Ge) çalışmalarında başarı oranı artmakta, Ar-Ge maliyetleri azalmakta ve Ar-Ge süresi kısalmaktadır (Viana ve ark., 2018).
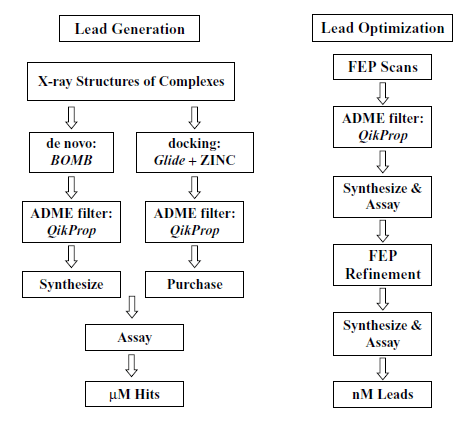
Bilgisayar destekli ilaç tasarımı programları ligand ve reseptörle ilgili bilgi birikimine ihtiyaç duyar; biyoinformatik araçlar, uygulamalar ve veri tabanlarına bağlı olarak gelişim gösterir. Bir hedefin (reseptör) varsa bunun ligandı ile birlikte 3 boyutlu yapısının (x-ışını veya NMR ile) bilinmesi gerekir; hiçbir deneysel veri yoksa, sekans verilerinden yola çıkarak homoloji modellemesi ile hedef molekülün 3 boyutlu yapısı oluşturulmaya çalıştırılır.
İlaç tasarımında iki temel yaklaşım vardır: ligand-tabanlı ve reseptör tabanlı moleküler tasarım metotları.
Ligand tabanlı yaklaşım genellikle hedef molekülün yapısı hakkında yeterli bilgi yoksa tercih edilir. Aktivite gösteren ligand ya da ligandlar model alınarak bunlar üzerindeki gerekli modifikasyonlarla molekül aktivitesi ve ilaç olabilirlik özellikleri artırılabilir. (Lavecchia ve Cerchia, 2016) Günümüzde genel aktivite tarama testleri, daha rasyonel ligand tabanlı yaklaşım olan kantitatif yapı-etki ilişkisi analizi (Quantitative Structure-Activity Relationship Analysis, QSAR) yöntemiyle birlikte yürütülmektedir.
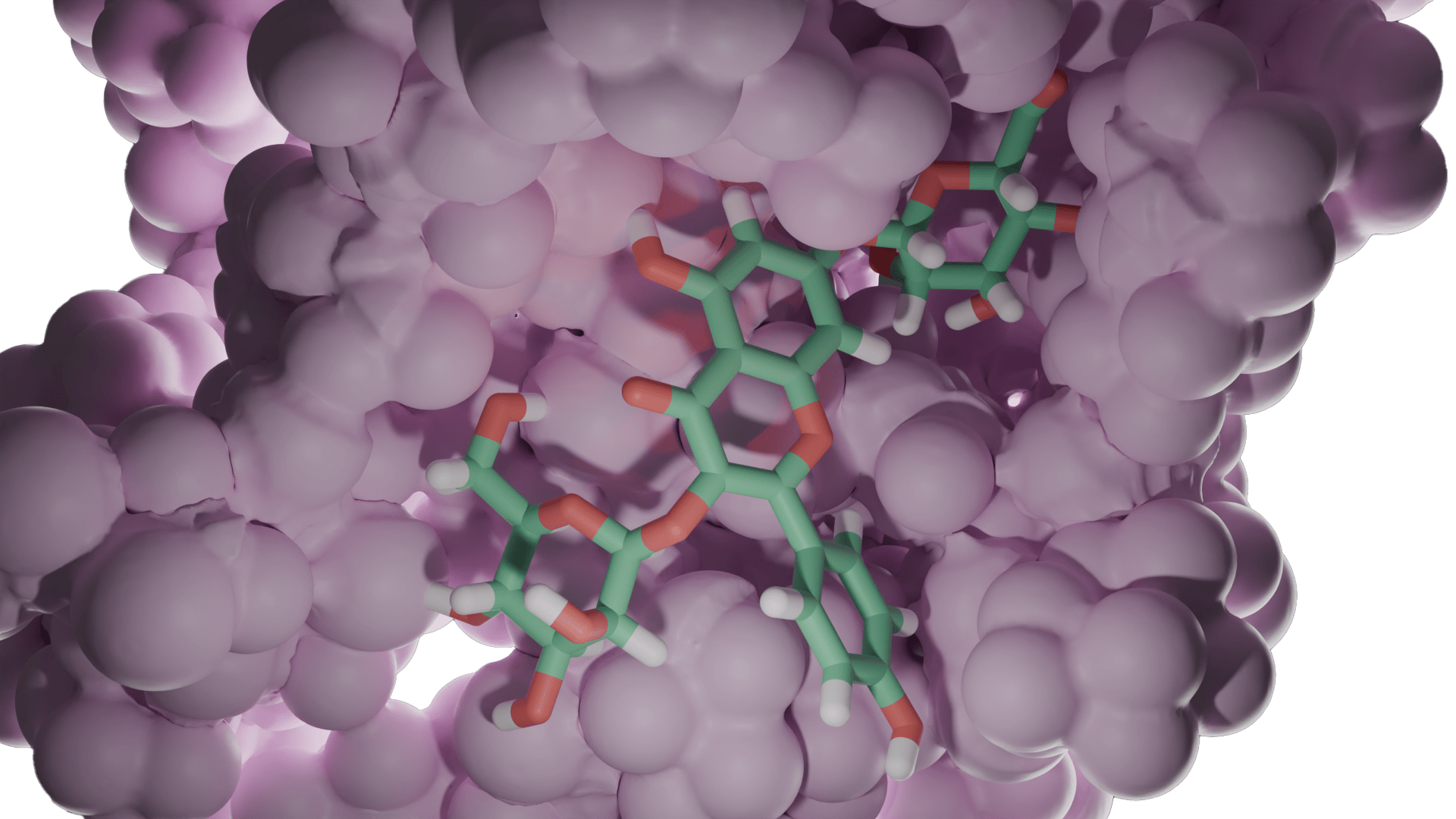
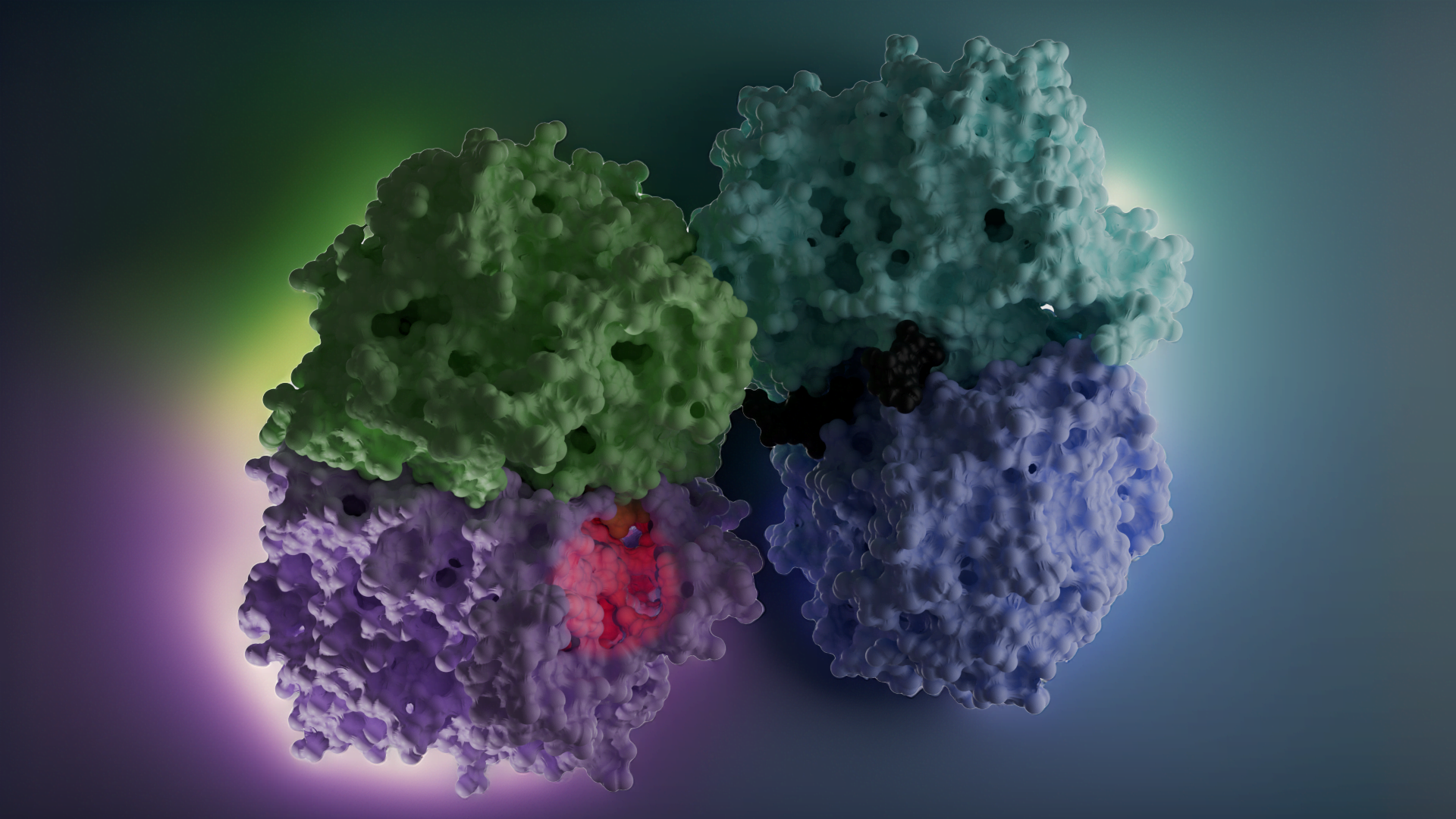
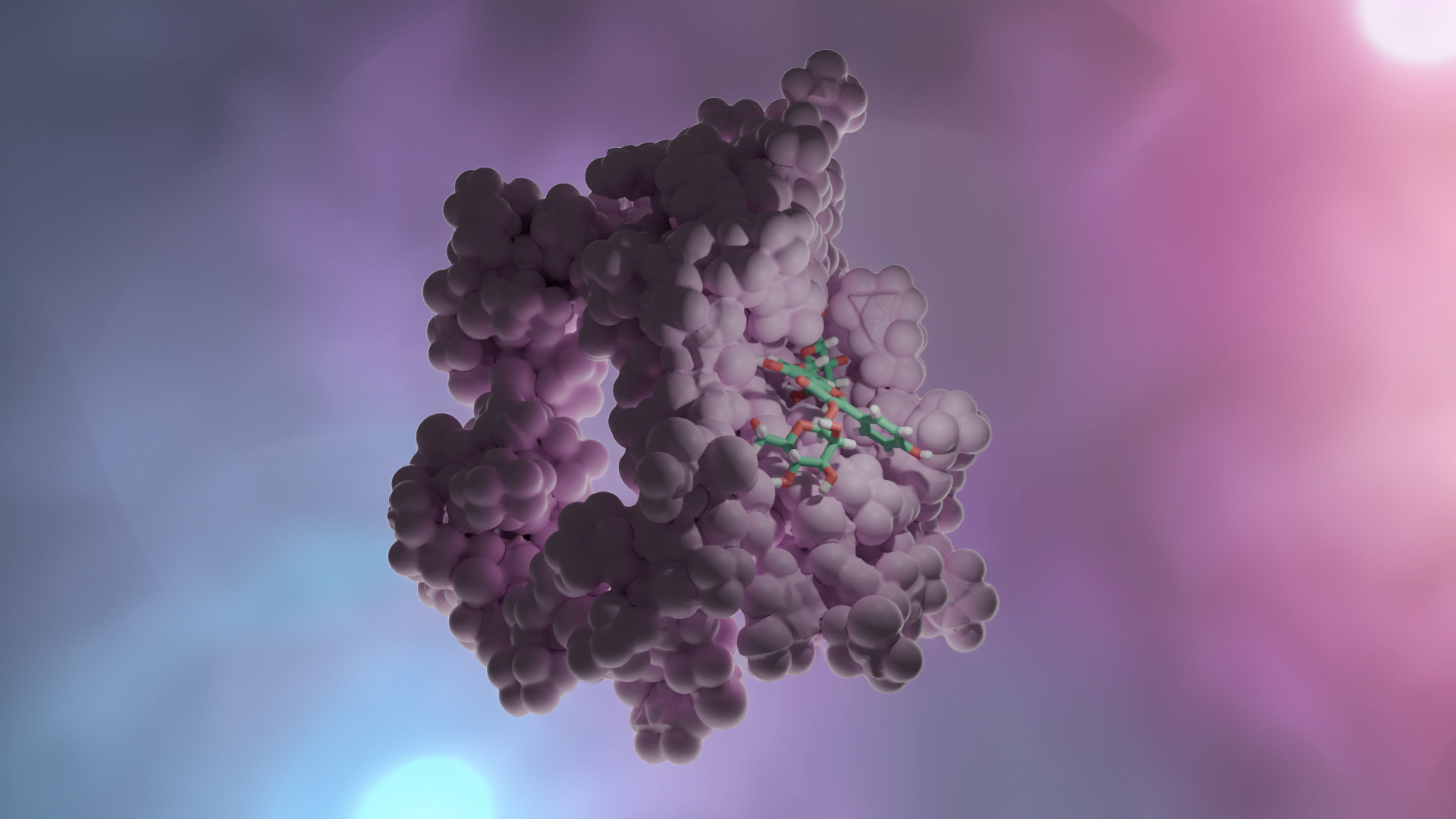
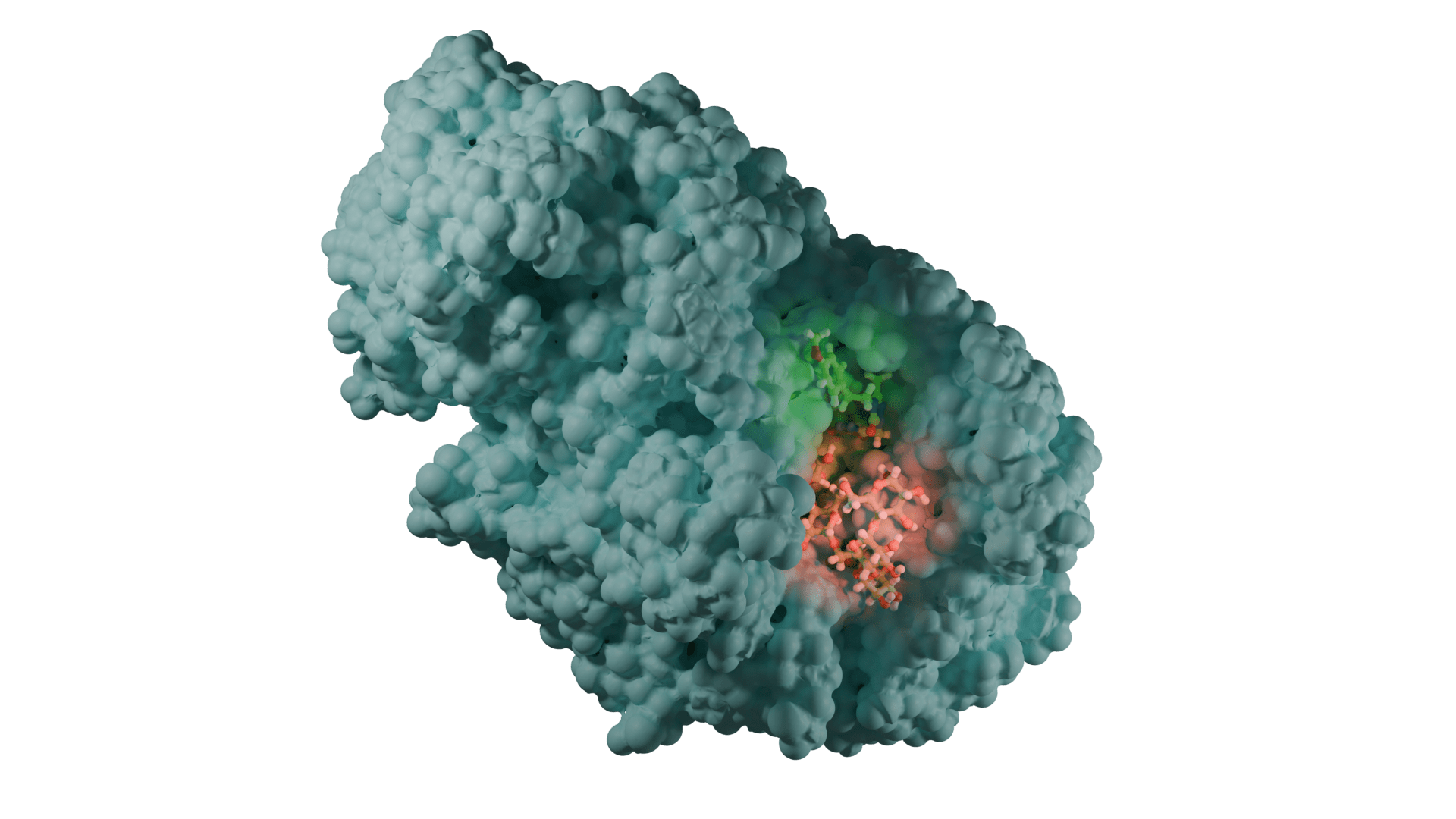
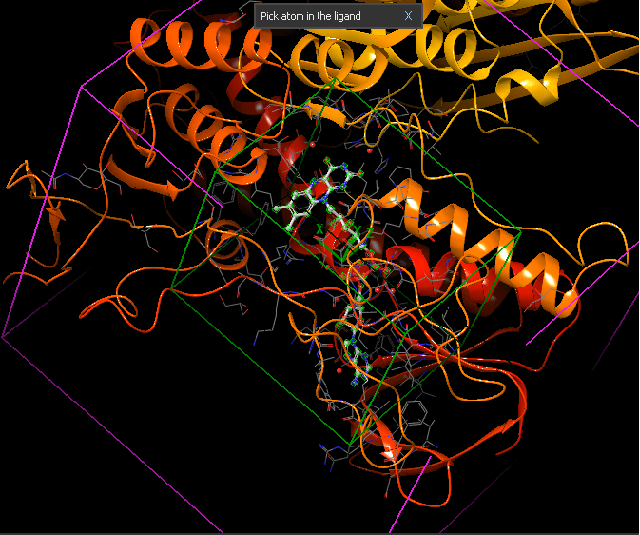
Yapı (reseptör)-tabanlı yaklaşım ise hedef molekülün yapısı hakkında güvenilir verilerin olduğu durumlarda tercih edilir (Daina ve ark., 2019 örneğin kristal yapısı aydınlatılmış bir enzim PDB: 3DKS). Hedefle ilgili 3B yapı elde edildikten sonra (X-ışını, NMR, homoloji modelleme ile) ligandların hedefin aktif bölgesi ile etkileşmesi docking hesaplamaları ile belirlenir. Bir hedef ile ligand arasındaki optimum etkileşme ilacın aktivitesinin artırılacağı ya da yan etkilerini azaltacağını garanti etmez. Ayrıca bu yaklaşım ilacın farmakokinetiğini de dikkate almaz.
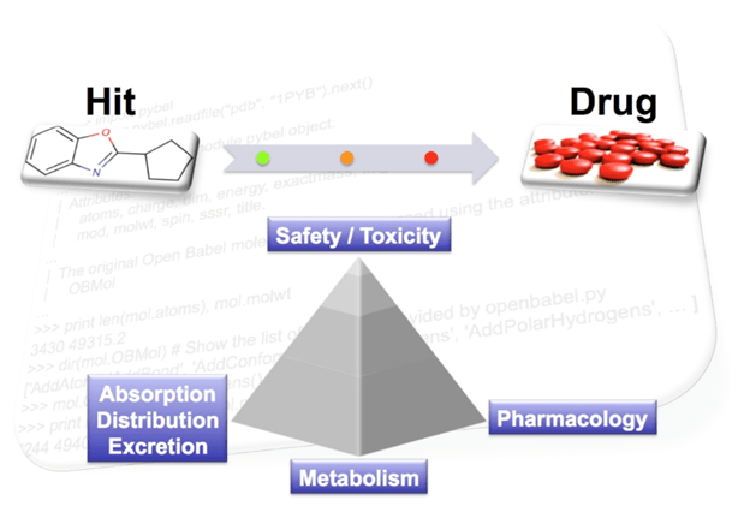
Yapı-temelli ilaç tasarımı tekrarlamalı (iteratif) bir yöntemdir. Önce hedef proteinin yapısı (x-ray, NMR, veya homoloji yöntemi ile) belirlenir. Çeşitli algoritmalar kullanılarak, veri bankalarından elde edilen bileşik ya da bileşik fragmanları (parçaları) yapının seçilen bir bölgesinde pozisyonlandırılır. Bu bileşikler hedef ile etkileşimlerine göre skorlanır ve derecelendirilir. Bu aşamada Moleküler Dinamik Simülasyonları ile daha net veriler eldd edilebilir. En iyi skora sahip bileşikler biyokimyasal deneylerle test edilir.
Bilgisayar Destekli İlaç Tasarımı için Diğer Adımlar
Yüksek verimli ilaç tarama (High Throughput Screening, HTS) ve bunun in silico muadili olan, protein-ligand docking tekniklerine dayanan, Sanal Tarama (Virtual Screening, VS) öncül ilaç keşfinde sıklıkla kullanılan yöntemlerdir. Bu yöntemlerde temel amaç belirli bir molekül kütüphanesi içinden en iyi sonuçları veren yüksek skorlu bileşiklerin, yaygın ifadesiyle “hit moleküllerin” belirlenmesidir. VS yönteminin bir zayıflığı “hit”leri belirlerken sınırlı bileşik veri-tabanlarından yararlanmasıdır.
Bilgisayar destekli ilaç tasarımı ile ilgili örnek çalışmalara bakmak için Moleküler Docking ve İlaç Tasarımında Kullanımı ve Raltegravir – İntegraz İnhibitörlerinin İlki yazılarımıza bakabilirsiniz. Bu yazılarımızda şuan çeşitli hastalıkların tedavisi için kullanılan ilaçların nasıl tasarlandığı adım adım anlatılıyor.
Bilgisayar destekli ilaç tasarımı alanlarından biri olan de novo ilaç tasarım yöntemi ile gerekli farmakolojik profilleri daha çok taşıyan yüksek verimli yapı iskeleleri ve moleküller oluşturulabilir. Çok sayıdaki ilaç-benzeri kimyasallar dikkate alındığında, de novo ilaç adaylarının belirlenmesi için daha uygun bir yöntem olabilir. Docking-temelli VS programlarıyla karşılaştırılırsa, de novo ilaç tasarım programı ile çok daha az sayıda “hit”ler ve öncüller geliştirilmiştir. Güncel de novo ilaç tasarım programında önemli bir problem, bu algoritmalar tarafından üretilen moleküllerin çoğunun sentezlenme güçlüğüdür.
Bilgisayar destekli ilaç tasarımı ile ilgili daha ileri çalışmalar moleküler dinamik çalışmalarıdır. Bu çalışmalarda kenetlenme çalışmalarının analizleri daha sağlıklı olarak yapılmaktadır. Bu çalışmaların özelliği tüm yapıdaki atomların her birinin bir diğer atom ile olan etkileşimini hesaba katarak sonuç üretmesidir. Moleküler dinamik çalışmaları ile ilgili daha çok bilgi almak ve bilgisayar destekli ilaç tasarımı konusunu daha iyi kavramak için Moleküler Dinamik Simülasyonları adlı yazıyı okuyabilirsiniz.
İlaç Yan etkilerinin dünya geneli istatistiksel raporlarına ulaşmak için ConDrug İlaç Güvenliği Platformuna üye olabilirsiniz https://condrug.com/
Videolara Göz At! Artık Her Şey Hareketli! Yeni Bilgiler Edin! Moleküler Dinamik Simülasyonları Bilimin Önemli Konularından.