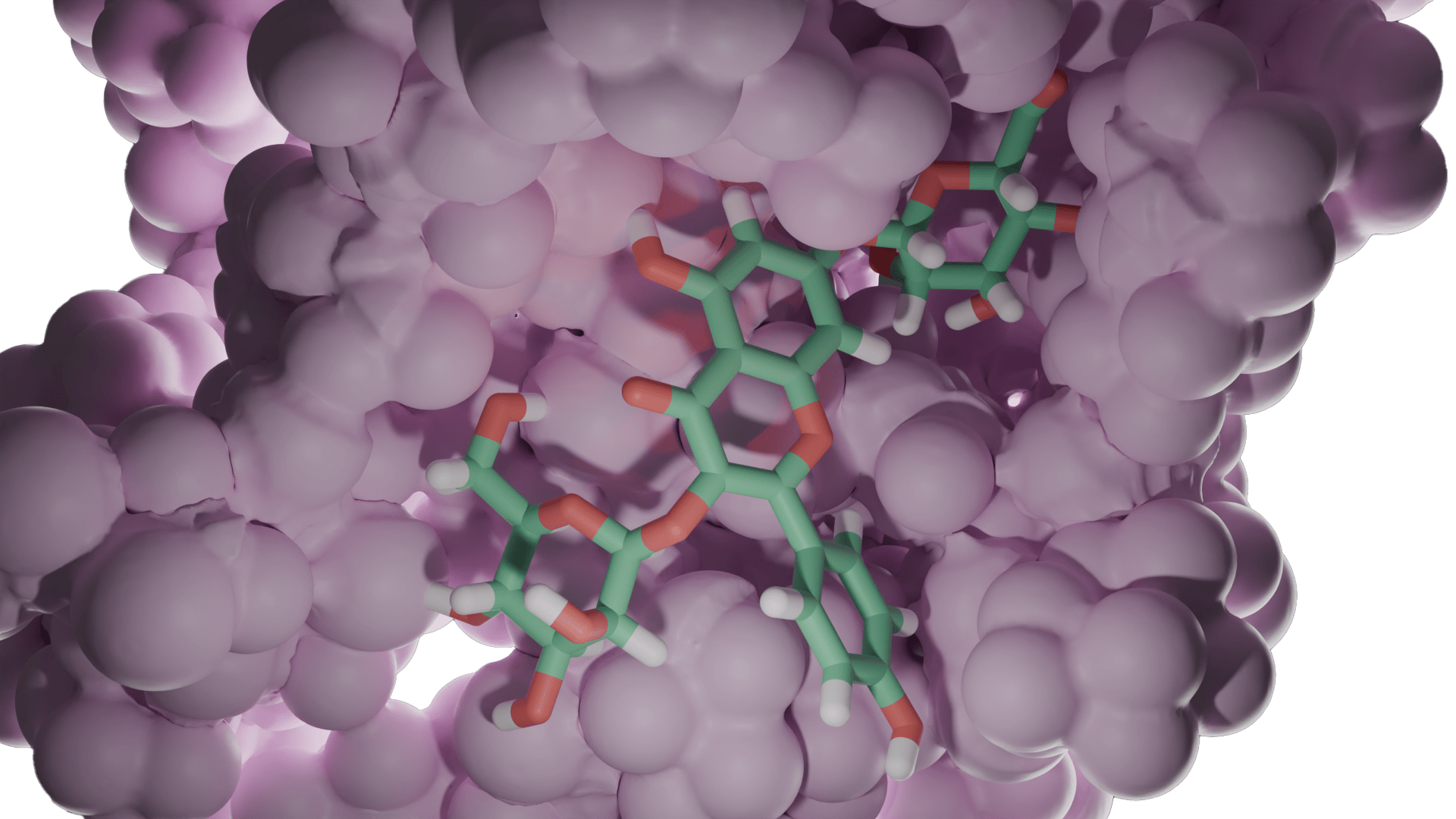
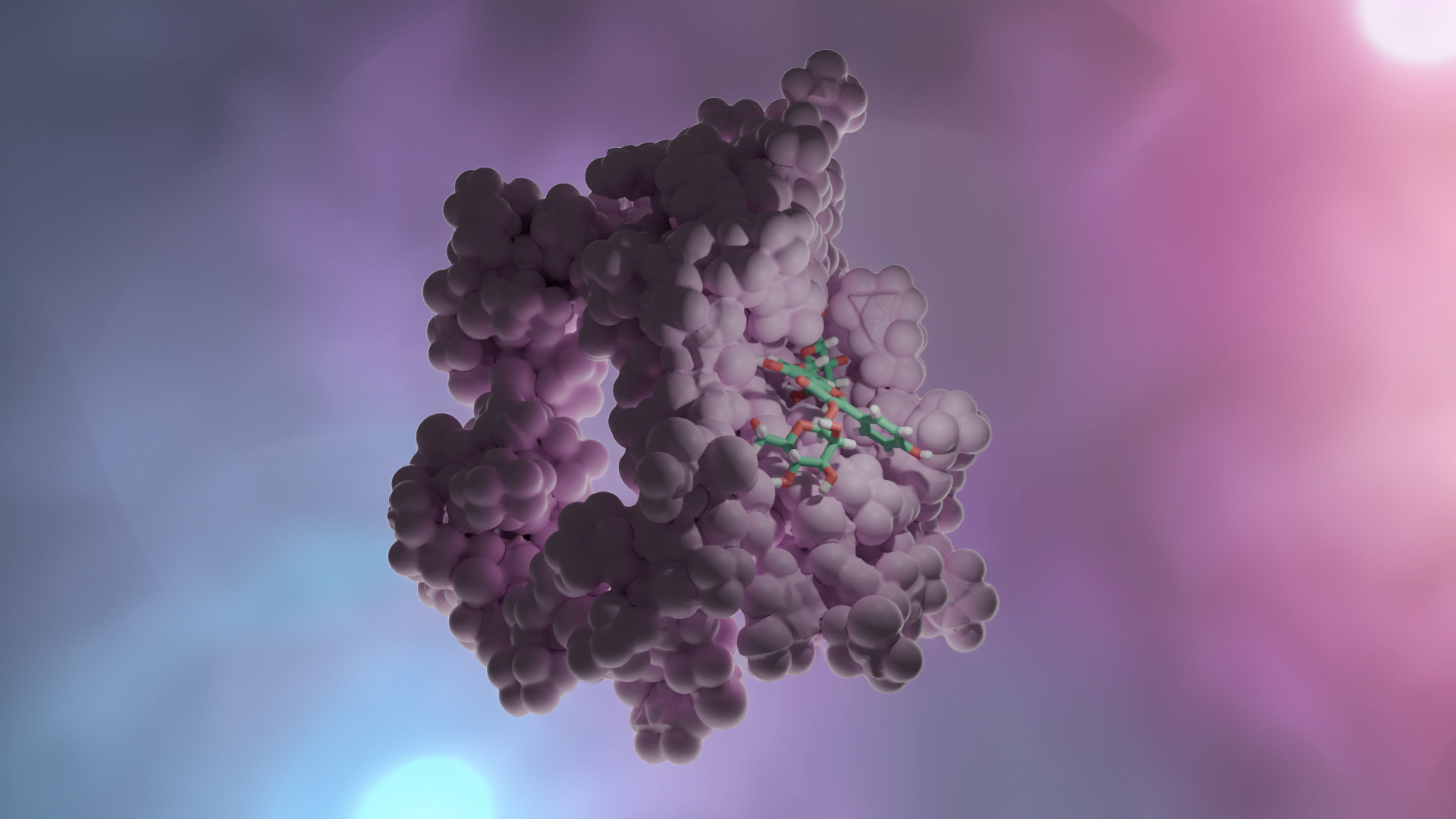
Moleküler docking, bir hedef ve ligand arasındaki etkileşim enerjilerini ve bağlanma afinitelerini hesaplayarak öngören bir yöntemdir. Rijid reseptör docking yönteminde, hedef proteinin aktif bölgesindeki amino asit yan zincirleri hareketsiz tutulur, içindeki ligand hareketlidir; en uygun kenetlenmeyi sağlamak için döner, titreşir ve bükülür; hedefin amino asit yan zincirleri ile non-kovalent etkileşimler yapar. Induced Fit Docking (IFD) gibi docking programlarında, hedefin aktif merkezinin bir miktar (bir kaç angström) hareket etmesine izin verilebilir [Schrödinger Suite 2019-1].
Tipik bir moleküler docking çalışmasında, biyolojik hedefin aktif bölgesi, 3D koordinat sisteminde ayrı ayrı parsellere (gridlere) ayrılır. Bu aktif bölgedeki atomların kısmi yükleri ile grid boyunca gezdirilen ligand molekülü arasındaki etkileşim enerjileri hesaplanır [Mügge and Enyedy (2004)]. Bu veriler daha sonra, her docking yönteminde farklı algoritmalar kullanılarak, toplam etkileşim enerjisine dönüştürülür. Bu işleme “skorlama” adı verilir. Bu skorlama fonksiyonudan elde edilen etkileşim enerjisi değerine “docking skorlama” denir. Tüm docking çalışmaları benzer işlemleri yapsa da, skorlama fonksiyonları birbirinden farklıdır. Bir ilaç keşfinde, docking ve skorlama fonksiyonları araştırmacıların ilgi alanlarına, protein ve / veya ligandlardaki atom tiplerine ve taranacak molekül sayısına göre seçilir.
Bir ligandın hedef ile kenetlenmesinde önemli rol oynayan hidrojen bağları, lipofilik ve iyonik etkileşimler, dönme enerjisi değişimi skorlama fonksiyonuna ilave edilmelidir. Ancak, genel docking algoritmasında [Böhm (1994)] bu terimler, zaman kazanmak için genellikle sıfırdan hesaplanmak yerine deneysel olarak belirlenmiş sabitlere dönüştürülür. Bu dönüştürme işlemi, bu algoritmaları yarı deneysel hale getirir ve özellikle üzerinde çalışılacak binlerce bileşik varsa göz ardı edilmemelidir. 1994 yılında Böhm tarafından önerilen bir protein-ligand etkileşim enerjisinin hesaplanması için genelleştirilmiş bir formül aşağıda verilmektedir.
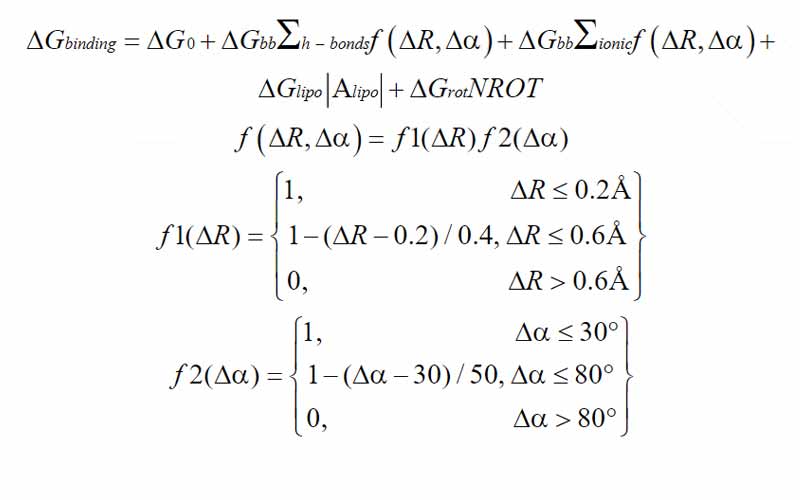
Moleküler Docking Algoritmaları
Söz konusu sisteme göre farklı yerleştirme ve puanlama algoritmalarını içeren piyasada bulunan birçok yerleştirme yazılımı vardır. Örneğin, hedef protein, yapısında, etkileşim haritalarında dikkate alınması gereken hetero atomlar (yani metal atomları) içerebilir. Veya hedef, hidrojen bağlarından ve helislerin genel biçiminden sorumlu fosfat gruplarını içeren ve daha fazla dikkat gerektiren bir DNA sarmalı olabilir. Araştırmacılar, hesaplamalar için doğru algoritmayı seçmek için incelenen sisteme dikkatle yaklaşmalıdır. HTVS de kullanılan docking etkileşim formülü şöyledir [Denklem 1.2].
Diğer Protokoller
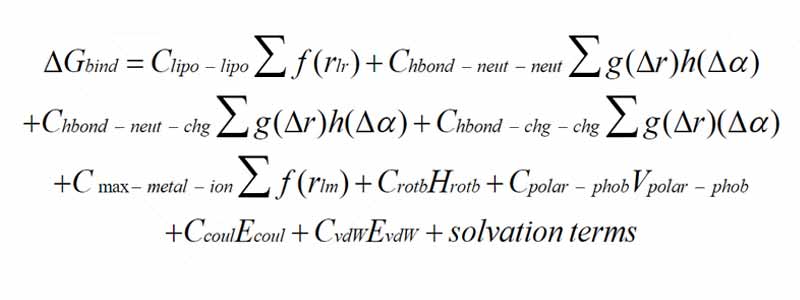
Ligand ve reseptör arasındaki lipofilik-lipofilik etkileşimler, protein ve ligandın nötr ve yüklü atomları arasındaki hidrojen bağı, metal ve ligand atomları arasındaki iyonik etkileşimler, polar ve hidrofobik atomların dönme enerjisi terimleri, elektrostatik ve van der Waals etkileşimleri, çözücü ve çözünmüş moleküllerden kaynaklanan çözülme enerjisi terimleri hesaba katılır.
HTVS ve SP yerleştirme protokolleri tarafından kullanılan puanlama işlevine ek olarak Glide XP, denklemlerine özel terimler getirmektedir. Bu yeni terimler, hedefler ve ligandlar arasındaki yapısal örüntüleri kapatarak ve daha önce bulunmayan veya birleştirilmiş su desolvasyon enerjisi terimlerini kullanarak bağlanma afinitelerinin geliştirilmesine yol açar. XP yerleştirme protokolüne dahil edilen ana geliştirme, bağlanma boşluğunun hidrofobik olarak kapatılmış bölgelerinin değerlendirilmesinin benzersizliğidir. 1.3 denkleminde görülebileceği gibi, bağlanma bölgesinin lipofilik ligand atomları ile nötr-nötr ve yüklü yüklü etkileşimleri içeren lipofilik alanlar arasında oluşan farklı tiplerde hidrofobik etkileşimler için birden fazla terim eklenmiştir. ve ligandın hesaba katılması üzerine su moleküllerinin bağlanma sahasında hariç tutulmasıyla ortaya çıkan desolvasyon terimlerini ve ayrıca ligand gerilme enerjilerini de içeren gelişmiş enerji cezası terimlerini geliştirmiştir. Glide XP algoritmasında kullanılan puanlama fonksiyonunun son şekli aşağıdaki gibi yazılabilir:

ADME özellikleri: Absorpsiyon, Dağılım, Metabolizma Ve Atılım
İlaç adayı çok sayıda molekül, çeşitli parametrelere göre incelenerek, hangi kimyasalların sentezleneceği ve test edileceği önceden belirlenir. Bir ilacın etkili olabilmesi için, yeterli derişimde vücuttaki hedefine ulaşmalı, biyolojik aktivitesi yüksek, toksisitesi (yan etki) düşük olmalı, biyoaktif formda yeterli bir süre bozunmadan kalabilmelidir. Molekülün farmakokinetik özelliklerini değerlendirmenin geleneksel yolu, hedefe erişimle ilişkili çeşitli etkileri farklı parametrelere ayırmaktır. Bunlar ADME parametreleri olarak bilinir. ADME parametrelerinin tahmini, moleküllerin klinik fazlardaki farmakokinetiğe bağlı başarısızlık oranını büyük ölçüde azaltmaktadır (Daina ve ark., 2017). ADME parametreleri, in siliko metodlar kullanılarak da belirlenebilir. Lipinski oral ilaçlarda gözlenen ortak fizikokimyasal parametreleri tanımlamış ve aralıklarını belirlemiştir. İlaç-benzerlik testi olarak bilinen Lipinski‟nin Beşli kuralı, farmakokinetik ve fizikokimyasal parametreler arasındaki ilişkiden yola çıkarak ilaç adayı bileşikte aranan yapısal özellikleri belirler (Daina ve ark., 2014).
1) molekulde hidrojen bağ donor atom sayısı sayisi 5‘ten fazla olmamalı (N-H ve O-H)
2) molekuldeki hidrojen bag akseptör atom sayisi 10’dan fazla olmamalı ( N ve O )
3) molekül kütlesi 500 dalton’un altinda olmali
4) lipofilite katsayisi (logP) 5‘in altinda olmali
Schrödinger platformu altındaki QikProp progamı ile ADME/Tox parametreleri hesaplanabilmektedir.

İlaç Yan etkilerinin dünya geneli istatistiksel raporlarına ulaşmak için ConDrug İlaç Güvenliği Platformuna üye olabilirsiniz https://condrug.com/
Videolara Göz At! Artık Her Şey Hareketli! Yeni Bilgiler Edin! Moleküler Dinamik Simülasyonları Bilimin Önemli Konularından.